问题来源:
利用MACS进行Peak-calling分析后继续进行motif-analysis,MEME-Chip的输入文件格式要求是fa文件,但是MACS产生的对应文件是bed格式,所以需要将bed文件转成fa文件
解决方法:
1.twoBitToFa
下载地址:
http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/
在linux上安装好后
输入
chmod 744 twoBitToFa (会将文件变成可执行文件,怎么实现的我也不懂)
再执行
twobittofa hg38.2bit hg38.fasta
或者
twoBitToFa -bed=xxxx.bed/leofs/noncode/reference/human/hg19/hg19.2bit out.fa #其中,xxxx.bed为需要转换的文件,/leofs/noncode/reference/human/hg19/hg19.2bit为人类参考基因组hg19.2bit的存储路径,out.fa为输出的文件(可根据需要命名成xxx.fa)
或者
twoBitToFa -bed=T.changhai_ALL_withinIntron_IGR_sigNC.bed ~/zhaogg/rna-seq-data/h19_bowtie_db/genome_all.fa_db.2bit 51T.fa
以上的执行命令都是我搜集到的(百度:如何将bed文件转换成fa文件)
但是不幸的是,并没有什么用(如果你会的话请告诉我 谢谢!)
2.bedtools
或许你会问:“博主你是不是脑子有问题,直接告诉我能使的不就行了么?”
“聪明,但是我是一个偏文科的理工科生,所以我习惯了先抑后扬。”
安装:
wget https://github.com/arq5x/bedtools2/archive/v2.26.0.tar.gz
tar -xvf v2.26.0.tar.gz
cd bedtools2-2.26.0
make(编译时间有点长)
代码:
bedtools getfasta -fi test.fa -bed test.bed -fo test_out.fa
#test.fa是参考基因组 test.bed是要转换的bed文件 test_out是输出的fa文件

ps:一定要在/bedtools2-2.26.0目录下执行
原理:
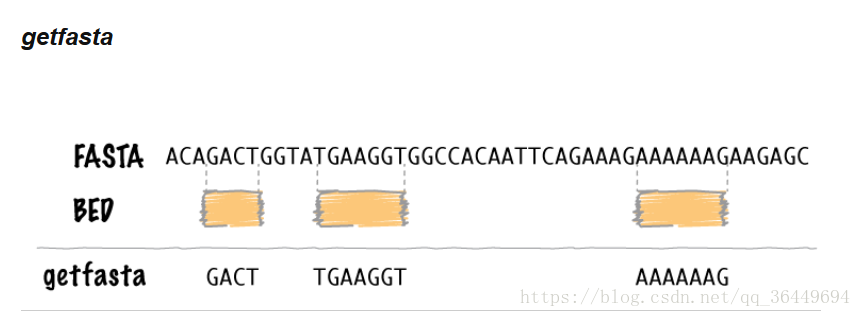
搞定收工~~