近日,Genomics, Proteomics & Bioinformatics 在线发表了中国科学院北京基因组研究所(国家生物信息中心)章张研究员题为“ KaKs_Calculator 3.0: Calculating Selective Pressure on Coding and Non-coding Sequences ”的应用说明文章。我们的“要文译荐”栏目很高兴邀请到章张研究员为大家解读KaKs_Calculator 3.0的研究情况。
摘 要
KaKs_Calculator是分子序列选择压力计算的工具包,最新版本3.0可支持编码和非编码序列的选择压力计算。编码序列的选择压力利用非同义替换率与同义替换率的比值来表征;与之类似,非编码序列的选择压力可用非编码序列的替换率与非编码序列临近编码序列的同义替换率的比值来计算。通过对真实数据的比较分析发现,KaKs_Calculator 3.0可准确计算出非编码序列的选择压力,从而为在全基因组水平上自然选择压力的解析和潜在功能原件的识别提供了重要方法。KaKs_Calculator 3.0工具包可免费获取用于学术研究。
KaKs_Calculator 3.0工具包免费获取网址: https://ngdc.cncb.ac.cn/biocode/tools/BT000001 长按并识别二维码 免费获取KaKs_Calculator 3.0工具包 |
|

引 言
自然选择压力的检测对分子进化、比较基因组学和系统发育重建具有重要意义,可揭示分子序列的进化过程及基因组演化的复杂分子机制。选择压力的理论计算需要一组不受选择影响的参考集合。同义替换不引起氨基酸的改变,因此被广泛用于中性进化的参考集。故而,非同义替换率(用Ka 或dN 来表示)与同义替换率(用Ks 或dS 来表示)的比值(Ka/Ks ),可用于鉴定中性突变、负选择与正选择,为编码序列的分子进化研究提供了重要的基础方法。
目前,越来越多的证据表明,非编码序列在多种生物学过程中发挥重要的功能作用,并与人类疾病密切相关。与编码序列相比,尽管传统认为其保守性较低,但已鉴定出大量非编码序列在哺乳动物基因组中高度保守。尤为重要的是,大量研究表明,非编码序列的确受到正选择和负选择的影响,特别是长非编码RNA(long non-coding RNA, lncRNA)序列。为此,研究人员已提出了几种计算方法来检测作用于非编码序列的选择压力,其主要区别在于如何选取不受选择影响的参考集,例如相邻编码基因的同义替换、内含子序列或者是祖先重复序列。然而,至今仍缺乏相应的算法,来实现非编码序列选择压力的检测。更为迫切的是,需要一个能够检测编码序列和非编码序列选择压力的集成工具包,这将帮助用户实现对分子序列自然选择压力的全基因组检测。
为此,我们开发了KaKs_Calculator更新版3.0,实现编码序列和非编码序列的选择压力检测。不同于之前的版本都集中在编码序列选择压力检测,基于相邻编码序列的同义位点作为参考集,KaKs_Calculator 3.0设计实现了非编码序列的选择压力检测算法,通过利用真实数据进行测试,验证其在非编码序列的分子演化强度和模式的检测能力。
算 法
KaKs_Calculator 3.0的主要更新内容是加入了一种能够检测非编码序列选择压力的算法。具体来说,它使用同义替换作为参考,尽管有学者认为同义替换也受到弱选择影响,但其仍被广泛应用于编码序列选择压力的计算。与编码序列的Ka/Ks 类似,对非编码序列的选择压力(ξ)可以量化为非编码核苷酸替换率(Kn )与中性替换率(假设为Ks )的比值,ξ = Kn/Ks ,其中Ks 是基于相邻的编码序列计算得到。由于观察到的替换数量少于实际替换的数量,我们采用核苷酸替换模型(例如,JC/K2P/HKY)进行多重替换校正。以HKY模型为例,根据等式(1),Kn 可以从观察到的转换数和颠换数(分别为S和V)以及四种核苷酸频率(πA、πT、πG和πC)推导出来。
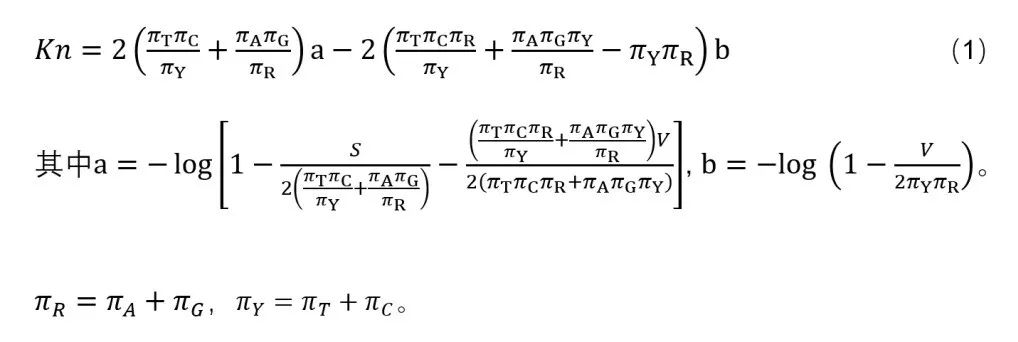
为了检测和量化非编码序列的选择压力,KaKs_Calculator 3.0为用户提供了两种获取中性突变率或者Ks 值的方法,该值可根据用户上传的相邻编码序列计算得出,亦可由用户直接输入指定(图1)。
图 1 KaKs_Calculator3.0的图形化用户界面
KaKs_Calculator 3.0采用标准C++语言实现,便于在不同操作系统(Linux/Windows/Mac)上的编译和高效运行。除了上述新功能外,新版本还修复了多处漏洞和错误。KaKs_Calculator 3.0工具包,包括已编译的可执行文件、图形用户界面(graphical user interface, GUI)的Windows应用程序、源代码和示例数据,以及详细的说明和文档,可在国家基因组科学数据中心(National Genomics Data Center, NGDC)的生信工具库BioCode上免费获取。
KaKs_Calculator 3.0工具包免费获取网址: https://ngdc.cncb.ac.cn/biocode/tools/BT000001 长按并识别二维码 免费获取KaKs_Calculator 3.0工具包 |
|

真实数据的应用
为测试KaKs_Calculator 3.0,依据LncRNAWiki知识库,我们选择了三个被广泛研究的lncRNA基因,并从NGDC LncBook和美国国家生物技术信息中心(National Center of Biotechnology Information, NCBI)收集它们的人鼠直系同源基因以及它们相邻的编码直系同源基因参考序列。具体来说,这些非编码基因和编码基因分别是:(1)H19 (NR_002196.2 vs. NR_130973.1) 和MRPL23 (NM_021134.4 vs. NM_011288.2) ;(2)Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) (NR_002819.4 vs. NR_002847.3)和SCYL1 (NM_020680.4 vs. NM_001361921.1);(3)Hox trascript antisense intergenic RNA (HOTAIR ) (NR_003716.3 vs. NR_047528.1) 和HOXC12 (NM_173860.3 vs. NM_010463.2) 。基于这些直系同源基因,我们通过MAFFT获得它们对应的比对序列(使用参数:--maxiterate 1000 --localpair)。
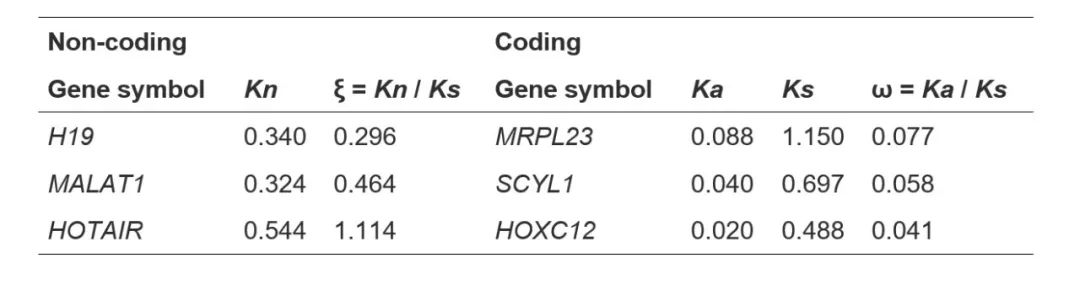
根据非编码核苷酸替换率与相邻同义替换率的比值(ξ),我们发现,尽管编码基因经历了强烈的纯化选择(ω < 1),这三个非编码基因呈现出不同的选择压力(表1)。值得注意的是,HOTAIR 表现出正选择(ξ > 1),而其余两个基因经历负选择(ξ < 1)。HOTAIR 是从HOXC基因簇的反义链转录而来的∼2.3-kb反义RNA。相对于HOXC12 ,HOTAIR 呈现出正选择进化,该结果与已发表文章的结论一致,即HOTAIR 比相邻基因进化得更快。MALAT1 是一种约8.7-kb的非编码RNA,侧翼是高度保守的激酶样基因SCYL1,在几乎所有人体组织中普遍表达,在哺乳动物物种中进化保守,并与各种癌症相关。因此,ξ = 0.464表明MALAT1 受强选择性约束,与其生理和病理功能及其保守的RNA结构相符合。同样,H19 是一种位于MRPL23 附近的约2.3-kb母体印记基因,它与Beckwith-Wiedemann综合征密切相关,并且参与肿瘤发生。结果表明,H19 呈现出更强的选择约束(ξ = 0.296),与其保守的序列和结构非常吻合。值得注意的是,一个非编码序列可能有多个相邻的编码基因,指定不同的编码基因会导致不同的Ks 和ξ。总之,KaKs_Calculator 3.0可有效检测非编码序列的自然选择压力,进而揭示编码序列与非编码序列的演化过程。
表 1 人-鼠直系同源基因选择压力与替换率估计
此外,为测试 KaKs_Calculator的运行性能,我们从NCBI RefSeq收集整理出含有15,424 个人鼠直系同源基因的大型数据集,并通过 ParaAT工具构建其编码序列的比对结果。KaKs_Calculator 3.0涵盖十种用于检测编码序列选择压力的计算方法,可分为:近似法和最大似然法。因此,我们选择三种常用的近似法NG(Nei and Gojobori)、YN(Yang and Nielsen)和 MYN(Modified YN),以及一种最大似然法GY(Goldman and Yang),并在4个CPU内核(单核3.4 GHz主频)的64位 x86 Intel Core i7并装载了Windows10的台式机上进行测试。对于这种大规模数据分析,我们发现NG、YN和MYN运行时间大概需要约2分钟,GY需要约11小时,表明近似法比最大似然法更省时。考虑到不同用户的使用偏好,需要注意的是,最大似然法理论上更为准确,同时不同的计算方法采用不同的模型和策略,因此可能会得到不同的计算结果。
讨 论
KaKs_Calculator 3.0实现了对非编码序列的自然选择检测。正如先验数据所证明的那样,它在计算序列的自然选择方面非常有用,从而可在全基因组范围内识别潜在的功能元件。未来的发展包括:检测非编码序列中小肽(少于300个核苷酸)的选择压力,整合基于密码子的比对流程以方便用户生成输入序列。
文章编译来源:Zhang Zhang. KaKs_Calculator 3.0: Calculating Selective Pressure on Coding and Non-coding Sequences. Genomics Proteomics Bioinformatics 2022. doi: 10.1016/j.gpb.2021.12.002. 英文全文详见:https://www.sciencedirect.com/science/article/pii/S167202292100259X.
GPB论文: KaKs_calculator 3.0: Calculating selective pressure on coding and non-coding sequences 长按并识别二维码 阅读原文 |
|

相关推荐
About GPB
Genomics, Proteomics & Bioinformatics(基因组蛋白质组与生物信息学报,简称GPB)于2003年创刊,是由中国科学院主管、中国科学院北京基因组研究所(国家生物信息中心)与中国遗传学会共同主办的英文学术期刊,由Elsevier金色开放获取(Gold Open Access)出版。刊载来自世界范围内组学、生物信息学及相关领域的优质稿件。现为中国科学引文数据库(CSCD)和中国科技论文与引文数据库(CSTPCD)核心期刊,被SCIE、PubMed / MEDLINE、Scopus等数据库收录。2018—2021连续位于中科院文献情报中心期刊分区表大类“生物1区Top"。2020年,CiteScore为12.4,位于“计算数学”、“遗传学”、“生物化学”、“分子生物学”四个学科领域Q1区;2年和5年Impact Factor分别为7.691和11.12,位于“遗传学与遗传性”学科领域前10%。期刊由科技部等七部门联合实施的“中国科技期刊卓越行动计划”资助(2019–2023)。
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
文献阅读 热心肠 SemanticScholar Geenmedical
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
点击阅读原文