一、遍历文件
设置工作目录的路径:
setwd("D:/file")
查看当前工作目录:

获得该目录下扩展名为txt的文件名:
temp=list.files(path="D:/file",pattern="*.txt")
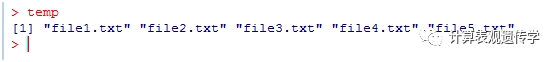
如果想读取其中一个文件,比如说file2.txt:
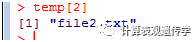
file=read.table(temp[2],sep='\t')
如果想批量处理文件,则可设置for循环读取处理:
for(i in 1:length(temp)){
file=read.table(temp[i],sep='\t')
}(注:此处代码仅是遍历读取,若是想批量处理文件,在循环内写入所需命令)
二、批量输出文件
设置工作目录路径,以便批量输出文件到该目录下:
setwd("D:/result")
查看当前工作目录:

批量设置输出文件名:
name=paste("result",c(1:5),".txt",sep="")

如果前缀想改成不一样的名,如果输出文件少则:
name=paste(c("h","e","l","m","o"),".txt",sep="")

如果输出文件多,则需将文件名放入列表后处理:
name=paste(na,".txt",sep="")

for循环批量输出文件:
for(i in 1:nrow(file)){
re=file[1,]
write.table(re,name[i],quote=FALSE,col.names=FALSE,row.names=FALSE,sep='\t')
}
三、遍历文件+批量输出文件
当需要一边遍历文件一边输出文件时:
设置工作目录的路径:
setwd("D:/file")
temp=list.files(path="D:/file",pattern="*.txt")
name=paste(c("h","e","l","m","o"),c(1:5),".txt",sep="")
for(i in 1:length(temp)){
file=read.table(temp[i],sep='\t')
index=which(as.double(file[,3])>1)
file_1=file[index,]
setwd("D:/result")##设置输出路径
write.table(file_1,name[i],quote=FALSE,col.names=FALSE,row.names=FALSE,sep='\t')
setwd("D:/file")##更改路径为原来的工作目录,方便循环
}
往期「精彩内容」,点击回顾
DNA测序历史 | CircRNA数据库 | Epigenie表观综合 | 癌症定位
BWA介绍 | 源码安装R包 | CancerLocator | lme4 | 450K分析
乳腺癌异质性 | BS-Seq | 隐马模型 | Circos安装 | Circos画图
KEGG标记基因 | GDSC | Meta分析 | R线性回归和相关矩阵
精彩会议及课程,点击回顾