测序知识
萱_1014已关注
0.1552020.06.21 17:10:13字数 1,784阅读 74
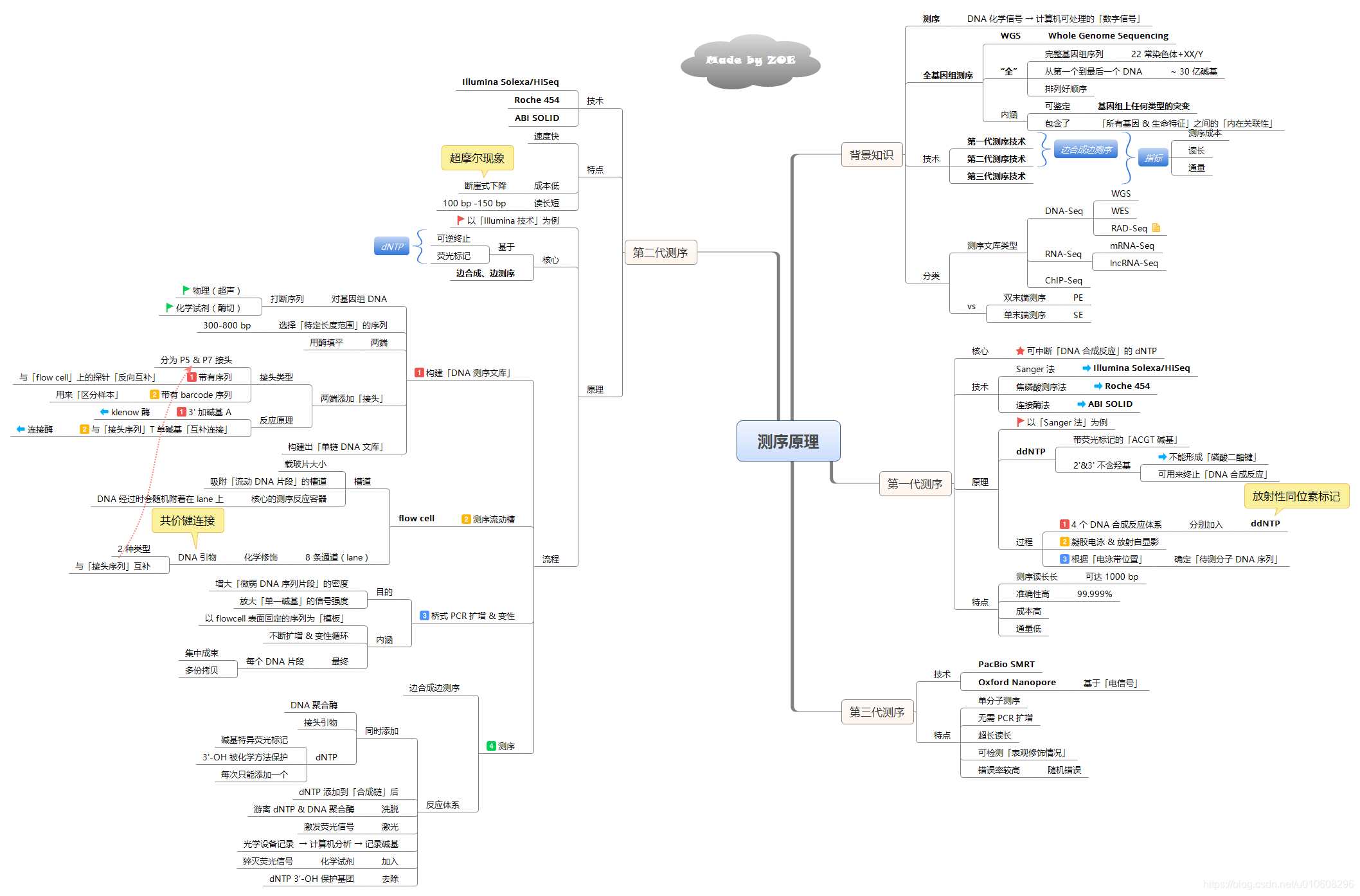
测序过程和原理
测序原理:
一代测序(Sanger测序):
(1)目前一代测序在验证序列(就是平时送公司测序返回来自己blast的那些)以及验证基因组组装完整性方面都是金标准。
(2)一代最长能测1000bp,
(3)它一次只测一条,也就是所谓的通量低
(4) 准确度很高,99.999%
NGS (next generation sequencing )二代测序,又称下一代测序:
名词:
flowcell: 测序反应的载体/容器,1个flowcell有8个lane
lane: 测序反应的平行泳道,试剂添加、洗脱等过程的发生位置
tile: 每次荧光扫描的位置,肉眼是看不到的
双端测序: 可能序列比较长有四五百bp,两边各测120-150bp
junction: 双端测序中间一些没有测到的区域
flowcell构造:一个lane包含两列(swath),每一列有60个tile,每个tile会种下不同的cluster,每个tile在一次循环中会拍照4次(每个碱基一次)
边合成变测序(sequence by synthesis, SBS)~合成
第一步: 构建DNA文库
第二步: 上样
三步:桥式PCR
第四步:测序
数据初步分析:
使用fastqc进行质量分析,这是一款Java软件,支持多线程
软件前期准备:下载方式有两种:
1.官网 http://www.bioinformatics.babraham.ac.uk/projects/fastqc/下载好用filezilla导入linux服务器
2. 直接在服务器中wget http://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.7.zip
接着安装unzip fastqc_v0.11.7.zip --> cd FastQC -- > chmod755 fastqc (这一步开始报错,找不到chmod命令,用conda安装后,还是找不到,which chmod,发现chmod在bin/下,目前还未解决这个问题)
设置完权限后,还需要将FastQC文件夹(这里请注意是文件夹,而非fastqc这个可执行程序)导入环境变量
echo 'export PATH=/YOUR/FASTQC PATH/:$PATH' >> ~/.bashrc
再source ~/.bashrc检查软件是否安装成功 fastqc --help 出现帮助信息就可以使用啦!
二代测序大幅度提高了测序速度,降低了测序成本,保持了高准确性。缺点是读长短,拼接困难,pcr技术增加了测序的错误率.
三代测序(TGS)
第三代测序技术以PacBio公司的SMRT和Oxford Nanopore Technologies 的纳米孔单分子测序技术为标志,不需要经过PCR扩增,超长读长,可达二代测序的100倍以上,实现了对每一条DNA分子的单独测序。错误率比二代要高,达到10-15%。
三代测序错误随机,平均读长8 ~25kb ,相对于二代而言,测序成本偏高,同时测序错误率偏高。

名词优化结构
1.基因组学(核酸序列分析)
(1)全基因组测序(WGS)
(2)全外显子组测序(WES)
(3)简化基因组测序(RRGS)
①RAD-Seq ②GBS ③2bRAD ④ddGBS(也就是ddRAD)
作用:(1)基因组作图(遗传图谱、物理图谱、转录本图谱)(2)核苷酸序列分析
(3)基因定位(4)基因功能分析
其它: 以全基因组测序为目标的结构基因组学;以基因功能鉴定为目标的功能基因组学
2.转录组学(基因表达分析)
(1)mRNA-Seq
(2)IncRNA-Seq(长链非编码RNA)
(3)sRNA-Seq(主要是miRNA-Seq)
作用:
(1)获得物种或者组织的转录本信息(2)得到转录本上基因的相关信息,如基因结构功能等
(3)发现新的基因(4)基因结构优化(5)发现可变剪切(6)发现基因融合 (7)基因表达差异分析
3.蛋白质组学
(1)蛋白质组数据处理、蛋白及其修饰鉴定
(2)构建蛋白质数据库、相关软件的开发和应用
(3)蛋白质结构功能预测
(4)蛋白质连锁图
4.代谢组学
(1)代谢物指纹分析
(2)代谢轮廓分析
1. 第一代测序技术
特点:读长长(1000 bp),准确性高(99.999%),通量低。
2. 第二代测序技术
(1)DNA文库构建 (2 ) 簇的生成——桥式PCR (3)测序(4)数据产出
特点:通量高、时间短、读长短。
3. 第三代测序技术
单分子实时DNA测序。
(1)PacBio 实时单分子测序
(2)Complete Genomics公司的复合探针-锚定连接技术
(3)Oxford Nanopore 纳米孔单分子通道技术
(4)Ion Torrent电子流检测技术
Pacific Biosciences公司的SMRT技术: SMRT测序速度快(每秒约数个dNTP),但是,测序错误率也较高(达到15%,可通过多次测序进行有效的纠错)。
Oxford Nanopore Technologies公司的纳米孔单分子技术为主流。
常用数据格式
1. Fastq格式
一种基于文本的,保存生物序列(通常是核酸序列)和其测序质量信息的标准格式,一般都包含有4行。
第一行:由‘@’开始,后面跟着序列ID和可选的描述,序列ID是唯一的;
第二行:碱基序列;
第三行:由‘+’开始,后面是序列的描述信息;
第四行:第二行序列的质量评价(quality value)。
Fasta格式:
(1)以“>”为开头,fasta格式标志。
(2)序列ID号,gi号,NCBI数据库的标识符,具有唯一性。
格式为:gi|gi号|来源标志|序列标志(接收号、名称等),若某项缺失可以留空,“|”保留。
(3)序列描述。
(4)碱基序列,序列中允许空格、换行、空行,一般一行60个。
格式见间的转化:Fastq文件→Fasta文件
Linux命令
法1:sed '/^@/!d;s//>/;N' your.fastq > your.fasta
法2:seqtk seq -A input.fastq > output.fasta
FASTX-Toolkit
一款用于处理Short-Reads FASTA/FASTQ文件的程序,里面包含了丰富的Fasta/Fastq文件格式转换、统计等命令。
http://hannonlab.cshl.edu/fastx_toolkit/
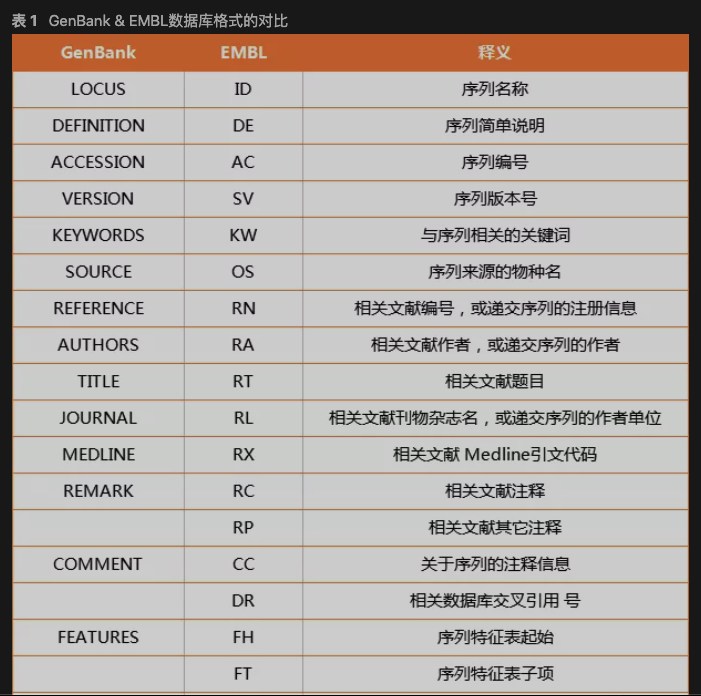
GenBank格式
以LOCUS和一些注释行开始。序列的开头以“ORIGIN”标记,末尾以“//”标记
EMBL格式
以标识符行(ID)开头,后面跟着更多注释行。
序列的开头以“SQ”开头标记,序末尾以“//”标记。
EMBL → Fasta格式转换(在线工具):
http://www.geneinfinity.org/sms/sms_embltofasta.html
另外给大家介绍一个常见测序文件格式解析的网站:
https://genome.ucsc.edu/FAQ/FAQformat.html#format1 (包含了各种各样的测序文件格式说明)

3人点赞