一、Metascape简介
Metascape(http://metascape.org/) 是一个功能强大的基因功能注释分析工具,能帮助用户将当前流行的生物信息学分析方法应用到批量基因和蛋白质的分析中,以实现对基因或蛋白功能的认知。只需在Metascape网页几步简单的操作,就可以对大批量的基因或蛋白质进行注释、富集分析以及构建蛋白质-蛋白质互作网络。并且构建的蛋白互作网络还可以直接导出给Cytoscape使用,绘制美观、可发表的蛋白互作网络图。
- 更新快:每月更新一次,保证了数据的可靠性;
- 覆盖广:整合了GO、KEGG、Uniprot等多个权威的功能数据库;同时Metasacape不仅可以分析人类(H. sapien)的数据,还包括很多其他物种数据,如 M. musculus, R. norvegicus, D. rerio, D. melanogaster, C. elegans, S. cerevisiae, A. thaliana, and P. falciparum等;
- 易操作:“CAME”流程操作,简单易上手,不仅可以单独分析一个基因集,还可以同时分析多个基因集;得到的结果报告颜值极高,往往能达到发表文章的级别!
- 不收钱!!!
2019年3月4日,Matascape团队总结了Metascape的使用就发表了一篇Nature communication,可见这个软件的被认可程度。其中提到Metascape网站在文章发表之前就已经被350多篇论文引用,其中不乏《自然》,《科学》,《细胞》等杂志。有趣的是约三分之二的引用文章直接使用了Metascape生成的图表。
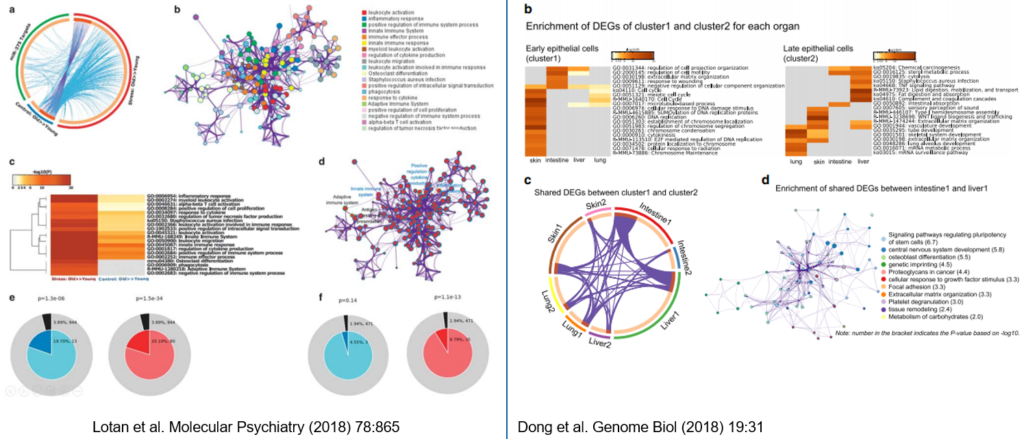 a)摘自Lotan et al. Molecular Psychiatry (2018) 78:865中的Figure 5; b)摘自Dong et al. Genome Biol (2018) 19:31中的Figure 3。
a)摘自Lotan et al. Molecular Psychiatry (2018) 78:865中的Figure 5; b)摘自Dong et al. Genome Biol (2018) 19:31中的Figure 3。
二、Metascape的工作流程
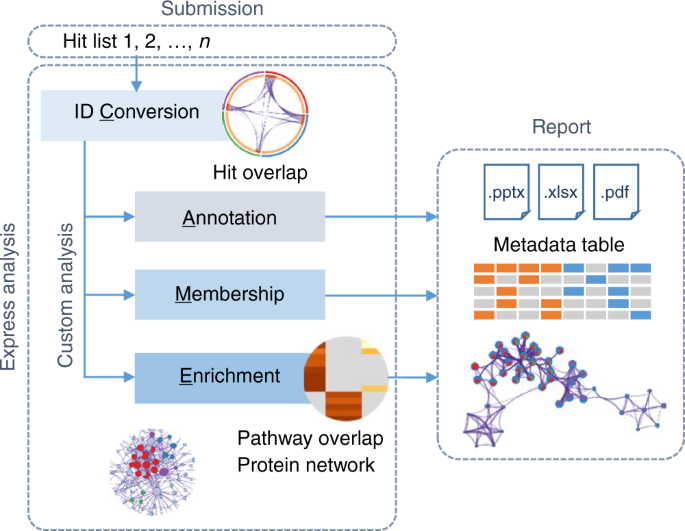
Metascape采用的是一种称为“CAME”的通用分析工作流程,概述如下:
-
ID转换 (Conversion)——将用户输入的基因标识符自动转换为目标物种的Entrez gene IDs;
-
注释 (Annotation)——为基因列表添加注释列,包括基因的描述、功能和蛋白质分类等;
-
归类 (Membership)——获取感兴趣的基因集。例如,获取被归为以“cancer”作为关键字的GO生物过程分类的基因。
-
富集 (Enrichment)——对基因列表的功能富集分析,识别具有统计意义的通路。最新的版本还包括蛋白质网络分析,可以用来识别和获取具有紧密连接的蛋白质网络元件 (如Oct4、Sox2和Nanog网络)。

新版本的Metascape可以分析多种模式生物的基因,用户只需输入基因或蛋白质名称 (很多基因名均可识别),就可以借助Metascape网站快速获得基因注释、基因功能、基因分类、富集的通路以及蛋白质相互作用网络(可导入Cytoscape再处理)。这对于大多数的日常分析需求来说已经足够了。
Metascape的使用
数据上传
在Metascape主页,用户可以在1处粘贴一个由逗号、冒号、空格、制表符或行分隔符组成的基因列表(基因名可以是:Entrez Gene ID、Ensembl ID、RefSeq、Symbol、UniProt ID、UCSC ID等等),或者在2可以选择本地的一个电子表格文件(xlsx、xls、csv或txt),其中的一列必须包含基因名称列。其它的数据列是可选的,在分析期间会被忽略。
图2 Metascape Home

-
如果在上传文件中提供了多个列,而基因只是其中的一列,注意使用下拉菜单确保正确选择含有基因名称的列。
-
txt格式基本上与csv格式相同,只不过前者使用tab作为字段分隔符;txt格式不支持任何非标准的规则。
-
Metacape会将分析后的基因注释等结果加在输入基因的列表后面。
-
使用Excel表时需要注意,Excel文件虽非常常用,但其有时不能很好地引用基因symbols,因为某些基因symbols可能被错误地转换成日期和数字。例如“Mar1”、“Marc1”和“March1”都可能被Excel转换为3月1日,而基因符号“201E9”则可能被视为一个大整数。其他的例子如“1/6”、“12-14-90”、“2-Oct”、“9830125E18”等等,因此这些基因symbols不能被映射到Entrez gene IDs中,因而在分析过程中会被漏掉。为防止基因symbols被转换成日期或数字,可以用单引号来对符号进行前缀,例如,输入’Mar1。因此最好不要依赖于在xls或xlsx格式中使用基因symbols,而应使用诸如RefSeq之类的其他ID格式。Excel改变了你的基因名,30% 相关Nature文章受影响,NCBI也受波及 但是,如果使用.csv或.txt格式,就可随意使用基因symbols。
-
Metascape主页包含所支持格式的文件模板(在“Upload File Format”下,见图3);可以下载下来并依照这些例子载入数据。想要测试Metascape的运行,可以点击
single list,将上传一个人类基因列表。点击Test Identifiers下的任何链接,就会自动粘贴指定格式的列表基因ID。 -
注意:基因列的名称开头不能有下划线,下划线是为Metascape保留的。用户提供的列的名称的任何下划线都将被自动删掉。
图3 数据格式举例

富集分析
粘贴或上传好基因数据后,在图Step2中先选择Input as species,如果有对应物种的基因就选择对应物种,如果没有可以选择括号中数字最大的物种 (说明可以进行转换的基因比较多),选择Analysis as species也遵循上述原则。
选择好物种后点击Express Analysis即可进行快速分析。此时下方会显示一个进度条,之后会显示一个Analysis Report按钮,点击后会打开一个报告页面。

快速分析(Express Analysis)包括最流行的注释源和基因本体 (Gene ontology)分类 (结果如下图)。对于经验丰富的用户,或希望对分析选项有更大控制权的用户,可使用自定义分析Custom Analysis。设置其中的一些选项能够更好地控制CAME分析流程。
 a) Metascape去除功能冗余的富集通路,用简单明了的bargraph显示出最主要的实验结果;b)富集生物通路可以以网络方式表现,这更利于理解通路或生物过程之间的关系;c)Metascape自动抽取提交列表中蕴含的蛋白质互作用网络;d)为了更容易理解这一网络,Metascape采用成熟的MCODE算法寻找网络中的密集联结的蛋白质群,并对每个群的生物功能进行注释。
a) Metascape去除功能冗余的富集通路,用简单明了的bargraph显示出最主要的实验结果;b)富集生物通路可以以网络方式表现,这更利于理解通路或生物过程之间的关系;c)Metascape自动抽取提交列表中蕴含的蛋白质互作用网络;d)为了更容易理解这一网络,Metascape采用成熟的MCODE算法寻找网络中的密集联结的蛋白质群,并对每个群的生物功能进行注释。
分析结果
-
先看到的是如图5的富集总结,横坐标是对p-values取以10为底的对数值并取负值;纵向是不同的富集通路,已按照-log10§的值排序。越排在上面的-log10§值越大,p-values就越小,富集就越显著(颜色也越深)。
-
在富集的通路中包括CORUM、Ractome和GO等数据集,展示的结果直观、丰富。可点击下方的
PDF下载。
图5 Heatmap of enriched terms across input gene lists, colored by p-values.

- 在下图的表格中包含上图中富集分析的具体信息,如
Count(即用户输入的基因有多少个落在这个通路中)和Log10(q)(是经多重假设验证校正的p-value)。
图6 Pathway and Process Enrichment Analysis

蛋白蛋白互作
下图展示了在用户输入的基因中发现的蛋白——蛋白互作网络。其数据来源于BioGrid、InWeb_IM和OmniPath等数据库。点击CYS 图标可以下载这个网络并可以用Cytoscape软件打开,可以进一步调整图形布局或加入基因表达等信息。
- Cytoscape教程1
- Cytoscape之操作界面介绍
- 新出炉的Cytoscape视频教程
- Cytoscape制作带bar图和pie图节点的网络图
- Cytoscape: MCODE增强包的网络模块化分析
图7 Protein-protein Interaction Enrichment Analysis

多基因列表联合分析
当代的多组学实验往往生成多个基因列表,目前的网络工具很少能同时分析并整合多基因列表,而这恰恰是Metascape的长处之一。其实Metascape的”meta”就是来源于多列表的meta-analysis。下图以三组过去独立发表的流感宿主因子列表为例进行说明。
 a) Metascape用heatmap让三组数据集共享的和独特的生物通路一目了然。b)富集通路也可以以网络呈现。由于每一组宿主因子用一个独特的颜色表示,我们可以很清楚的发现
a) Metascape用heatmap让三组数据集共享的和独特的生物通路一目了然。b)富集通路也可以以网络呈现。由于每一组宿主因子用一个独特的颜色表示,我们可以很清楚的发现Viral gene expression是共享的而Regulation of cell development主要只存在于绿色对应的实验中。
作者自评
很多学者还在采用DAVID做富集通路分析。富集通路分析结果完全依赖于背后数据库的质量。DAVID曾经有六年的时间(2010-2016)没有维护数据库,最近的更新也已经两年半了。独立研究表明使用两年旧的Gene ontology数据库,用户平均要丢失20%的最新的生物知识。所以定期更新数据库的重要性非同小可。可惜现实是目前大家常使用富集分析工具中仅有40%被正常维护,不知读者目前使用的工具是否属于幸运的一类。Metascape每月更新背后的40多个数据库,以确保提供最准确的结果。
Metascape没有学习使用的壁垒,因为没有比一键Express Analysis更简单了。 虽然好用,Metascape实现的主要功能却是不折不扣。作者由于多年来在自己的科研项目中都要花大量的时间进行这些生物信息分析,所以决定把长期发表文章中体会到的best practices实现在Metascape中。由于常用的基因列表的分析工具仅限于提供单一通路富集分析,这就不幸给研究人员造成了列表分析就是等同于知识驱动的富集分析的误解。而数据驱动的蛋白质互作用网络分析就鲜有网站支持。其实除了以上描述的Metascape提供的林林总总的分析功能之外,其还有非常强大的对上千个基因进行注释或者利用知识库进行成员分析的功能,这些对于后续的基因筛选及其重要,详情请见文章或网站文档。要实现Metascape的这些分析功能对于生物信息人员都是有难度的。
生信宝典之傻瓜式
-
Metascape比GO分析更加简单易用,富集的内容和通路也更加丰富,并且可以绘制蛋白相互作用网络图;
-
相比DAVID,Metascape会定期更新数据(目前是每月更新一次)。并且所有数据源的最新更新报告可以在首页的“News”部分中找到。
-
Metascape的使用完全免费。
高通量测序分析会产生数以千计的候选基因,但是只有少部分的基因能用于下游分析。研究基因的功能是生物医学研究的重要课题之一。如果你关注的是一个基因,那么可以搜索该基因相关的文献或从WikiGenes等基因信息数据库中来快速了解这个基因。但如果关注的是多个基因,比如以下情形:
-
转录组数据中有显著差异表达的Top30基因集;
-
受一个或多个转录因子调控的一组基因;
-
受一个或多个miRNA靶向的多个基因;
-
受一个lncRNA顺式或反式调控的多个基因;
-
或者仅仅是你感兴趣的一个基因集。
那么该如何了解和分析一组基因的功能倾向?虽然基因每种功能都有相应的数据库,比如GO功能注释、KEGG通路注释、Uniprot蛋白注释等,但是我们不可能去每个数据库走一遍流程,尤其是当我们分析数百个基因或多个基因集的时候。