论文题目:Improved detection of DNA-binding proteins via compression technology on PSSM information
通过PSSM信息上的压缩技术改进DNA结合蛋白的检测
Abstract:
- 由于已经认识到DNA结合蛋白在多种生物分子功能中的重要性,越来越多的研究人员正试图鉴定DNA结合蛋白。
- 近年来,由于蛋白质序列数据的有利速度和准确性,机器学习方法在蛋白质序列数据飙升的情况下变得越来越引人注目。
- 在本文中,我们从蛋白质序列中提取三个特征,即NMBAC(归一化Moreau-Broto自相关),PSSM-DWT(位置特异性评分矩阵 - 离散小波变换)和PSSM-DCT(位置特异性评分矩阵 - 离散) 余弦变换)。
- 我们还在这些特征向量上使用特征选择算法。
- 然后,将这些特征作为分类器输入训练SVM(支持向量机)模型以预测DNA结合蛋白。
- 我们的方法应用三个数据集,即PDB1075,PDB594和PDB186,来评估我们的方法的性能。
- PDB1075和PDB594数据集用于Jackknife测试,PDB186数据集用于独立测试。
- 我们的方法在Jacknife测试中达到了最佳准确度,分别在PDB1075和PDB594数据集上从79.20%到86.23%和80.5%到86.20%。在独立测试中,我们的方法的准确率达到76.3%。
- 独立测试的表现也表明我们的方法具有一定的能力,可以有效地用于DNA结合蛋白的预测。
- The data and source code are at https://doi.org/10.6084/m9.figshare.5104084
Introduction
- DNA结合蛋白在多种生物分子功能中发挥重要作用,例如转录,DNA损伤和复制的检测。
- DNA结合蛋白的重要性促进了各种鉴定它们的方法的发展。
- 已用于鉴定DNA结合蛋白的实验方法包括滤膜结合分析,遗传分析,微阵列染色质免疫沉淀和X射线晶体学。然而,这些实验方法具有一些缺点,例如昂贵且耗时。特别是随着下一代高通量DNA测序技术的发展,蛋白质序列数据正在迅速增长。
- 为了进一步促进计算过程,已经开发了一些Web服务器来生成DNA,RNA或蛋白质序列的特征向量,例如称为Pse-in-One的Web服务器。
- 根据各种特征信息,基于ML的方法主要由基于结构信息和基于序列信息的方法组成。
- 蛋白质的结构特征与功能密切相关,因此基于结构信息的预测因子可以实现更好的DNA结合蛋白鉴定性能。
- Nimrod et al.使用蛋白质的平均表面静电势,偶极矩和基于簇的氨基酸保守模式训练随机森林分类器。艾哈迈德等人。 基于蛋白质的净电荷,电偶极矩和四极矩张量,开发了一种神经网络分类器。Bhardwaj等人。 利用SVM分类器和三个特征,包括表面和整体成分,整体电荷和正电位表面贴片。
- 一些基于结构的方法也具有序列信息的参与。Szila'gyi和Skolnick从以下三个角度提取特征向量:某些氨基酸的相对比例,某些其他氨基酸的空间分布的不对称性和分子的偶极矩。 然而,结构信息不能知道大量蛋白质,因此基于结构的预测因子只能应用于整个蛋白质数据库的一小部分。
- 相反,序列信息更容易提取并且更便于使用。
- 我们可以提取多种基于序列的特征,例如物理化学特性,二肽组成和氨基酸组成。
- Cai和Lin使用蛋白质的氨基酸组成训练SVM分类器,疏水性的有限范围相关性和蛋白质的溶剂可及表面积。Yu等人。 通过将这些特征(从蛋白质序列氨基酸组成和物理化学特性中提取)提供到SVM分类器中,开发了rRNA-,RNA-,DNA-结合蛋白的二元分类。刘等人。 从三个序列特征中提取特征向量,包括总氨基酸组成,假氨基酸组成和物理化学距离转换。
- 一些研究人员还将PSI-BLAST生成的进化信息整合到基于序列的方法中,以提高预测性能。库马尔等人。 他们是第一个使用进化信息来鉴定DNA结合蛋白并开发出一种称为DNA结合剂的SVM分类器。一些类似的方法,例如Ho等人的方法。 ,还提出了鉴定DNA结合蛋白。他们的结果表明,进化信息可以显着提高性能,因此进化信息可用于鉴定DNA结合蛋白。刘等人。 提出了一种名为iDNAPro-PseAAC的预测器,它包含进化信息和假氨基酸组成(PseAAC)。Waris等人的方法使用从二肽组合物,分裂氨基酸组成和位置特异性评分矩阵(PSSM)中提取的特征来训练多个分类器并找到实现最佳预测性能的分类器。
- 如上所述,特征提取算法确定蛋白质序列是否可以通过特征向量完全表达。为了获得满意的性能,我们应该仔细选择特征提取算法。
- 在本文中,我们创新地结合了名为PSSM-DWT的1040维特征向量,名为PSSM-DCT的100维特征向量和名为NMBAC的200维特征向量来预测DNA结合蛋白。离散小波变换(DWT)和离散余弦变换(DCT)可用于通过压缩PSSM矩阵来获得有效信息。此外,我们根据六种物理化学特性提取200维特征向量。将这些特征输入训练SVM模型以预测DNA结合蛋白。
- 我们通过三个数据集评估我们的方法,即PDB1075,PDB594和PDB186。前两个数据集用于Jackknife测试,最后一个数据集PDB186用于独立测试。
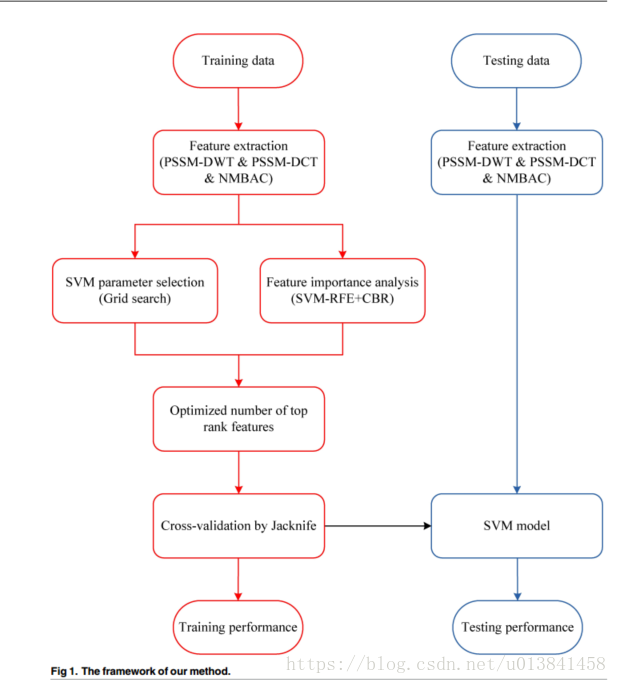
Materials and methods
- 在训练阶段,我们从PSSM矩阵中提取两个特征(PSSM-DWT和PSSM-DCT),并从六个物理化学特性中提取NMBAC特征。通过将这些特征馈送到SVM分类器中来获得预测模型。
- 在预测阶段,我们使用相同的特征表示算法来描述预测蛋白质序列,然后使用训练SVM模型进行DNA结合蛋白质预测。
Datasets
- 在本研究中,我们应用三个基准数据集来评估我们的方法,即PDB1075,PDB594和PDB186。这些DNA结合蛋白选自Protein Data Bank(http://www.rcsb.org/pdb/home/home.do)。
- 必须除去少于50个氨基酸或含有“X”字符的蛋白质序列。我们应该确保没有序列与任何其他序列具有超过25%的相似性。
- 具体地,由Liu等人构建的PDB1075数据集具有525个DNA结合蛋白和550个DNA非结合蛋白。由Lou等人编辑的PDB594数据集由297个DNA结合蛋白和297个DNA非结合蛋白组成。这两个数据集适用于Jackknife测试。 用于独立测试的PDB186数据集也来自Lou等人的论文,并且包含93个DNA结合蛋白和93个DNA非结合蛋白。
Evolutionary features进化特征
- Position specific scoring matrix.定位特定评分矩阵。(PSSM)
- 由PSI-BLAST生成的位置特异性评分矩阵(PSSM)(BLAST +选项:-num_iterations 3 -db nr -inclusion_ethresh 0.001)存储蛋白质序列的进化信息。Position Specific Scoring Matrix (PSSM) generated by PSI-BLAST [37] (BLAST+ [41] options: -num_iterations 3 -db nr -inclusion_ethresh 0.001)stores the evolutionary information of a protein sequence.
- 假设蛋白质序列的长度是L(L氨基酸),该蛋白质的PSSM的大小是L×20(L行和20列)。 该矩阵的形式如下:
- 每个元素PSSMoriginal(i,j)的公式如下:
- 其中ω(i,k)是位置i的第k个氨基酸类型的频率,D(k,j)是蛋白质中第k个氨基酸到第j个氨基酸的突变率 来自Dayhoff突变矩阵(替代矩阵)的序列。取代矩阵的值越大表明保守位置越强; 否则,相反。
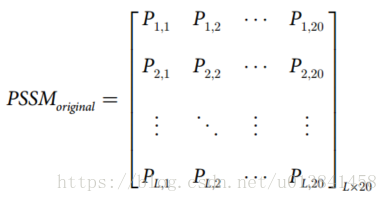
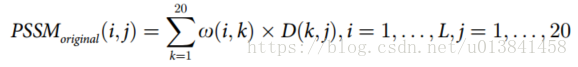
- 离散余弦变换。 我们使用离散余弦变换(DCT),它广泛用于数据压缩以压缩PSSM并将一部分压缩PSSM保留为特征向量。
- DCT是线性可分离变换,可以将信息密度的分布从均匀地改变为不均匀。压缩后,我们应保留PSSM的低频部分,因为低频部分包含的信息多于高频部分。在这项工作中,2维DCT(2D-DCT)用于压缩PSSM。 给定输入矩阵Mat = PSSMoriginal 2 <R L×20,相应的转换公式如下:
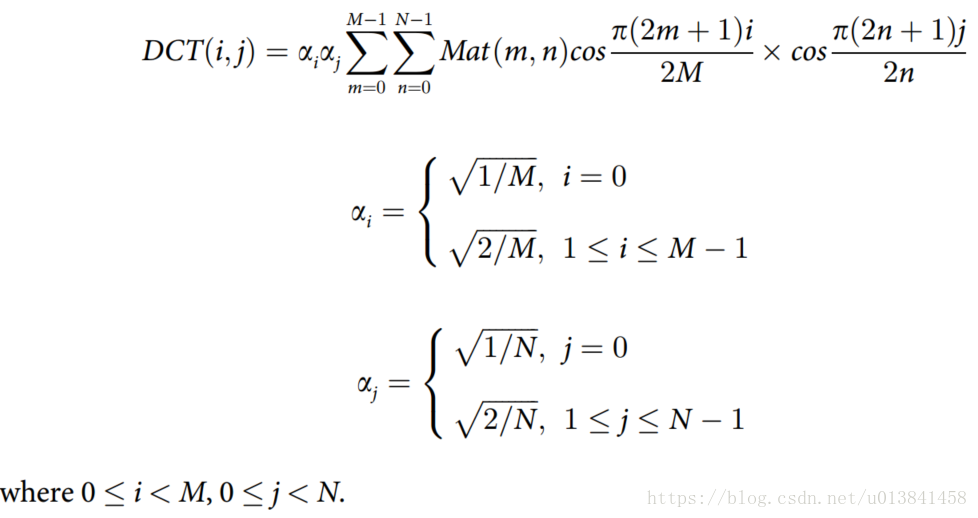
- 根据上述压缩公式,包含大部分信息的部分(低频部分)分布在压缩PSSM的左上角。
- 最后,我们将前100个系数保留为PSSM-DCT功能
- 离散小波变换。 小波变换(WT)定义为信号f(t)投影到小波函数上:
- 其中a是比例变量,b是翻译变量。
是分析小波函数。T(a,b)是为信号上的特定位置和特定小波周期找到的变换系数。
- 离散小波变换(DWT)可以将氨基酸序列分解为不同扩张的系数,然后从轮廓中去除噪声分量。
- Nanni等。 通过假设离散信号f(t)是x [n],提出了一种执行DWT的有效算法,其中N是离散信号的长度。
- 其中g是低通滤波器,h是高通滤波器。ylow [n]是信号的近似系数(低频分量)。yhigh [n]是详细系数(高频分量)。
- 重复该分解以进一步增加频率分辨率和用高通和低通滤波器分解的近似系数,然后进行下采样。
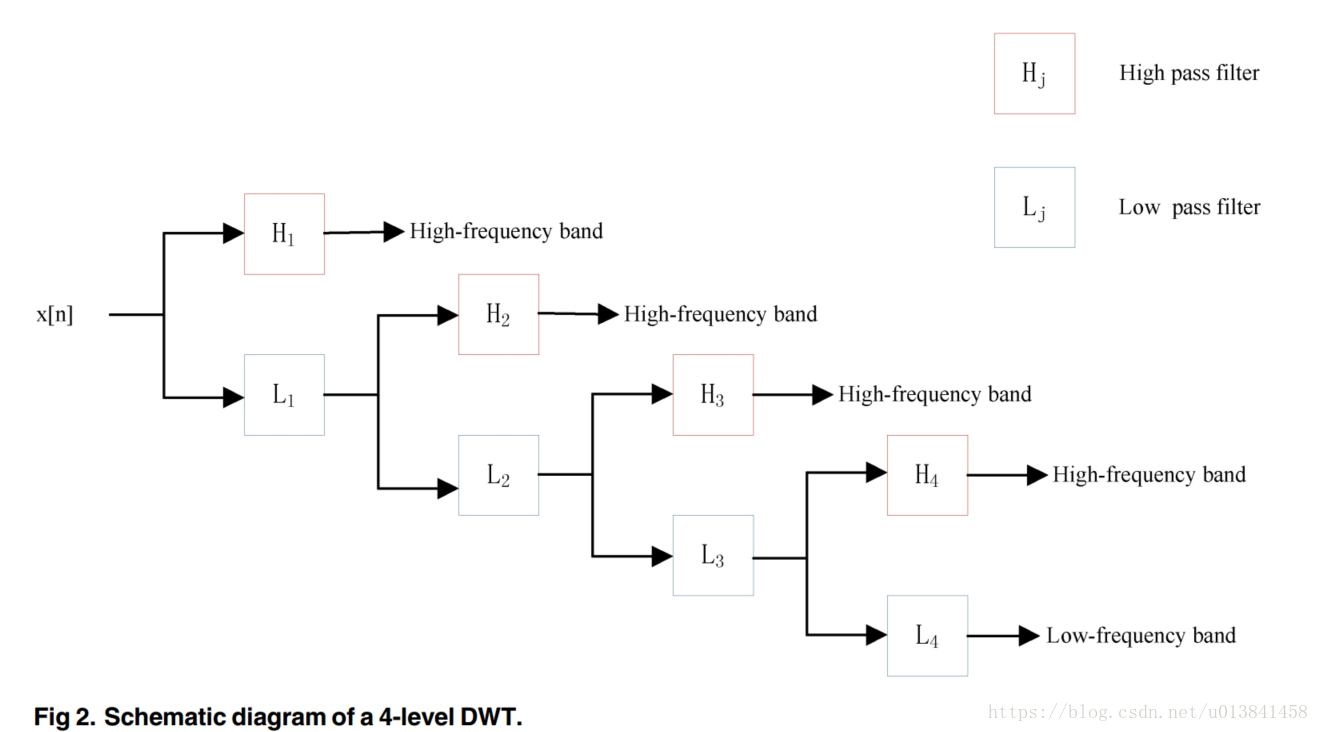
- 随着分解水平j的增加,可以观察到更详细的信号特征。受Nanni工作的启发,我们使用4级DWT并计算不同尺度的最大值,最小值,平均值和标准偏差值(低频和高频系数的4个等级)。
- 由于高频分量噪声较大,因此只有低频分量更重要,我们还从近似系数中提取前五个离散余弦系数。4级DWT的示意图如图2所示。
有20列。 因此,PSSM由20种类型的离散信号(L长度)组成。 最后,我们使用4级以上的DWT来分析PSSM(每列)的这些离散信号,并从蛋白质的PSSM中提取PSSM-DWT特征。
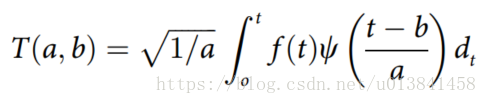

Sequence features
- 归一化Moreau-Broto自相关。我们使用归一化的Moreau-Broto自相关(NMBAC)从六种物理化学特性中提取序列特征以改善预测性能。
- NMBAC由Feng等人提出。 用于预测膜蛋白类型。20个氨基酸的每种物理化学性质具有相应的值,蛋白质序列可以用物理化学性质值的载体代替。
- 在我们的工作中,六种物理化学性质是疏水性(H),氨基酸侧链体积(VSC),极性(P1),极化率(P2),溶剂可及表面积(SASA)和侧链的净电荷指数 (NCISC)分别为氨基酸。20种氨基酸的理化性质值列于表1中。
- 在我们使用这些值来表示氨基酸之前,必须将它们标准化为零均值和单位标准偏差(SD),如下所示:
- 其中Pi,j是氨基酸类型i的描述符j的值,Pj是描述符值j的20个氨基酸的平均值,并且Sj是相应的SD。
- 对于每种物理化学性质,蛋白质可以由归一化的物理化学性质值组成的载体表示。
- 通过将这些向量输入以下公式获得NMBAC:
- 其中j代表六个描述符的一个描述符,i是蛋白质序列X中的位置,n是蛋白质序列的长度,lag是一个残基与另一个残基之间的连续距离,一定数量的残基(lag= 1,2) ,...,lg,lg是由要描述的优化过程确定的参数。

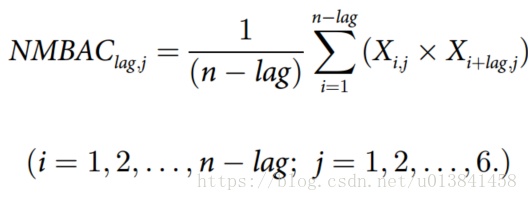
- 根据郭的工作,我们将滞后的最优值定义为1到30。对于每个蛋白质序列,我们可以获得30×6 = 180维特征向量。
- 我们还将该序列上出现的20个氨基酸的频率添加到特征向量中。最后,我们可以得到蛋白质序列的30×6 + 20 = 200维特征向量。
Classification and feature selection
- 在特征提取过程之后,基准数据集中的所有样本都被转换为具有相同维度的数字特征向量。
- 每个蛋白质序列的特征空间由PSSM-DWT,PSSM-DCT和NMBAC特征组成。
- 通过从原始特征空间(PSSM-DWT + PSSM-DCT + NMBAC)中移除噪声和冗余特征,特征选择可减轻过度拟合并提高性能。
- 为了减少特征丰度和计算复杂度,我们使用支持向量机递归特征消除和相关偏差约简(SVM-RFE + CBR)来选择最佳特征子集。 通过将CBR策略结合到特征消除过程中,提出了SVM-RFE + CBR:(1)不易过度拟合; (2)能够充分利用培训数据; (3)快得多,尤其是很多候选功能。
- 因此,它已成功应用于许多问题,特别是在基因选择方面。我们可以获得具有排名特征列表的SVM-RFE + CBR的输出。
- 通过选择一组排名靠前的功能来实现功能选择。 SVM-RFE + CBR的排序标准与SVM模型密切相关。
- 支持向量机。 由Vapnik开发的支持向量机(SVM)是一种分类和回归范例。
- 在使用SVM的过程中,使用内核将标记为正或负的样本投影到高维特征空间中,并且优化特征空间中的超平面以最大化正样本和负样本的边缘。
- 存在一些生物学问题,例如蛋白质 - 蛋白质相互作用的预测,同源性检测,以及可以使用SVM解决的基因表达数据的分析。
- 给定实例标签对{xi,γi}的训练数据集,i = 1,2,....。 。,N具有输入数据
和输出标签
,由SVM实现的分类决策函数用以下等式表示:
- 其中系数αi是通过求解下面的凸二次规划(QP)问题得到的:
- 其中xj被称为pupport向量仅当相应的αj> 0时,C是一个正则化参数,它控制边际和误分类错误之间的权衡。
- 在大多数情况下,称为径向基函数(RBF)核的
具有更好的边界响应,并且大多数高维数据通过类似高斯的分布来近似。我们使用具有径向基函数的LIBSVM [59]实现了SVM模型
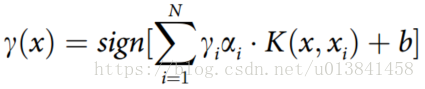
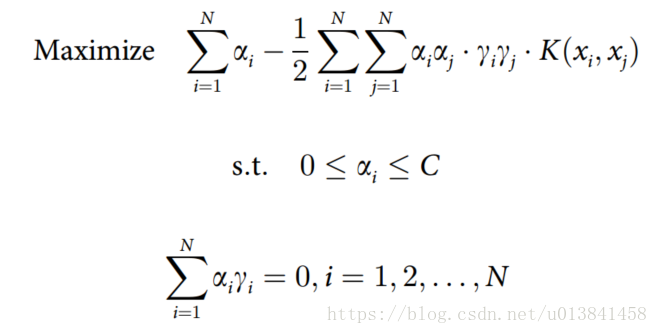
Results and discussion
扫描二维码关注公众号,回复: 3166983 查看本文章
- 我们在三个数据集上预先形成我们的方法来预测DNA结合蛋白。
- 在Jackknife测试中,我们将方法应用于PDB1075和PDB594数据集来分析特征提取和特征选择的有效性,我们的方法的性能也与其他方法进行了比较。
- 在独立测试中,我们的预测模型在独立数据集PDB186上进行测试,并与其他方法的结果进行比较。
Measurements测量
- 我们使用Jackknife测试来分析我们的方法构建的预测器的质量。由于Jackknife测试的有效性,它被广泛用于测试预测器的功能。
- 在Jackknife测试中,我们逐个使用基准数据集的每个样本作为测试数据集,其余样本用于训练预测变量。
- 此外,我们采用了四种方法,这些方法也用于其他方法来评估我们方法的性能,包括准确度(ACC),灵敏度(SN),特异性(SP)和马修相关系数(MCC)。 他们的公式如下:
- 其中TP是真阳性的数量,TN是真阴性的数量,FP是假阳性的数量,而FN是假阴性的数量。

Parameter optimization参数优化
- 为了选择特征NMBAC和PSSM-DCT的最佳参数,我们通过五重交叉验证测试不同参数(具有不同值lg的NMBAC和具有不同前m个系数的PSSM-DCT)的预测性能。
- 为了获得最佳lg,我们将lg的值从5到45评估(步长为5)。
- PDB1075数据集的预测结果如图3所示。当lg的值在5到30之间时,预测的ACC正在增加。之后,ACC的价值正在下降。
- m的不同值可能导致不同的性能,我们测试m的不同值从20到260(步长为20)。ACC的曲线如图4所示。当m从20增加到100时,ACC的值上升。
- 但当m在100和260之间时,它略有下降。显然,m小于100的PSSM-DCT(对于NMBAC小于30)将失去一些有效特征,较大的值可能会引入噪声。
- 因此,我们在实验中选择lg为30(NMBAC),m为100(PSSM-DCT)
Benchmark dataset—PDB1075
- 性能不同的功能。我们从基准数据集(PDB1075)中提取了三个特征,即PSSM-DWT,PSSM-DCT和NMBAC。
- 我们需要找到一组功能来实现最佳性能,并分析最重要的功能以获得良好的预测。
- 表2列出了Jackknife试验的不同特性。NMBAC,PSSM-DCT和PSSM-DWT的组合实现了最高的ACC(0.7926),MCC(0.5853),SN(0.8000)和第二高的SP(0.7855)。
- 为了获得每个特征的重要性,我们比较了通过Jackknife交叉验证在PDB1075数据集上获得的七个特征组合的AUROC,如图5所示。
- 我们可以看到对预测性能的最大贡献是PSSM-DWT,其次是NMBAC,而PSSM-DCT是最低的。
- 这些信息表明,每个特征都可用于预测DNA结合蛋白,并且三个特征的组合可以达到最佳性能,但PSSM-DCT特征不如其他两个特征有效。
Performance after feature selection.
- 为了提高PDB1075数据集的性能,我们通过SVM-RFE + CBR从原始特征空间中去除了噪声和冗余特征。
- 考虑三个特征的组合,首先,我们获得排名特征列表,如图6所示。NMBAC,PSSM-DCT和PSSM-DWT分为三个区间:[1,200],[201,300]和[301,1340]。
- 然后通过Jackknife测试得到的不同尺寸特征的精度可以在图7中看到,我们可以发现当我们选择第一个216维特征时可以获得最佳精度,这些特征可以从排名特征列表中获得:77个特征 在区间[1,200]中,区间[201,300]中的22个特征和区间[301,1340]中的117个特征。
- 特征选择应用于每个要素组合,我们可以获得具有每个要素组合的最佳性能的要素尺寸。
- 与前一节一样,我们还根据特征选择后七种特征组合的AUROC比较分析了每个特征的重要性,如图8所示。
- 我们得到的结果与NMBAC和PSSM-DWT在预测DNA结合蛋白方面比PSSM-DCT更有效。
- 对于上一节的结果,我们在特征选择后获得了新的结果,如表3所示。我们还发现,三种功能的组合可以实现最佳性能,并且在没有功能选择的情况下显然超出了性能。
- 它达到所有指标的最高值:ACC(0.8623),MCC(0.7250),SN(0.8743)和SP(0.8509)。 这些结果有力地证明了特征选择可以显着提高预测性能。
- 与现有方法比较。我们的方法在PDB1075数据集上的表现与其他现有方法进行了比较,包括iDNA-Prot | dis [40],iDNA-Prot [61],DNA-Prot [19],PseDNA-Pro [36]。 ,DNAbinder [38],iDNAPro-PseAAC [25],Kmer1 + ACC [23]和Local-DPP [62]。
- 表4显示了Jackknife测试的不同方法的性能。我们可以发现,通过我们的方法评估的四个测量值显着高于其他方法的评估测量值。与其他方法相比,我们方法的ACC,MCC,SN和SP值分别提高了7.03%,0.13,2.63%和4.73%。
基准数据集-PDB594
- 我们将我们的方法的性能与应用于基础数据集(PDB594)的Lou方法[2]的几个分类器进行了比较,如表5所示。
- 我们的方法达到最高ACC为86.2%,MCC为0.724,SN为87.2%,SP为85.2%。ACC,MCC,SN和SP值分别提高了5.7%,0.114,1.7%和7.1%。这代表了我们鉴定DNA结合蛋白的方法的有效性。
独立数据集-PDB186
- 为了分析鲁棒性,我们的方法与独立日期集(PDB186)上的其他方法进行了比较(PDB1075用作训练数据集,PDB186用作测试数据集),如表6所示。我们的方法达到了ACC的76.34%, MCC为0.5566,SN为92.5%,SP为60.22%。 我们的方法仍然比大多数具有一定可信度的现有方法表现更好。
计算时间
- PDB1075的特征提取和折刀测试评估的计算时间如表7所示。从表中可以看出,使用特征选择算法的折刀测试评估的计算时间明显短于折刀。没有特征选择的测试评估。 这证明了特征选择算法可以有效地减少冗余特征。
Conclusion
- 在本文中,我们提出了一种新的特征提取算法来构建DNA结合蛋白预测的机器学习方法。
- 我们采用特征提取算法提取三个特征向量,即NMBAC,PSSM-DWT和PSSM-DCT。我们应用DWT和DCT方法是有意义的,这些方法很少用于生物信息学以获得PSSM-DWT和PSSM-DCT。
- 通过这些方法,从PSSM矩阵中提取有效信息并将其存储在特征向量中。
- 在Jackknife测试中,我们的方法可以达到很好的预测性能,并且我们的预测性能在特征选择后明显超过了其他现有方法。在独立数据集上,我们的方法仍然比大多数现有方法表现更好。
- 此外,我们可以发现PSSM-DWT特征对预测性能的贡献最大。我们的方法的性能证明了特征提取算法的合理性和我们的方法在预测DNA结合蛋白的有效性。