前几天有同学问了一篇文章里的一个方法的实现,看了一下这篇文章除了
qPCR验证基本都是纯生信,今天就试着来复现一下。随缘复现哈,如果阅读数据不好看的话,可能就放弃了,希望大家多多点赞、在看,转发支持。

文章标题:Investigation of a Hypoxia-Immune-Related Microenvironment Gene Signature and Prediction Model for Idiopathic Pulmonary Fibrosis.
doi: 10.3389/fimmu.2021.629854
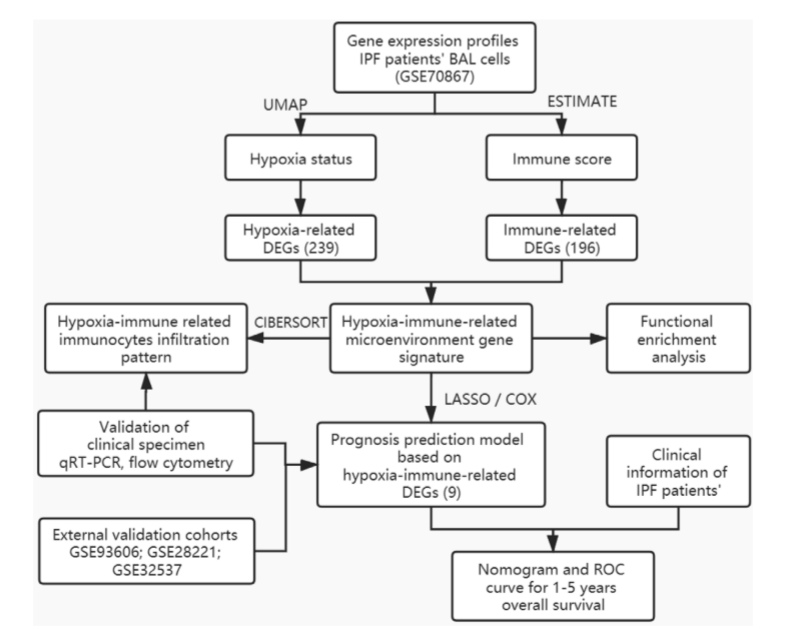
流程
示例数据和代码领取
点赞、在看本文,分享至朋友圈集赞10个并保留30分钟,截图发至微信mzbj0002领取。2022年VIP会员免费领取。
木舟笔记2022年度VIP企划
权益:
2022年度木舟笔记所有推文示例数据及代码(含大部分2021年)。

木舟笔记科研交流群。
半价购买
跟着Cell学作图系列合集(免费教程+代码领取)|跟着Cell学作图系列合集。
收费:
99¥/人。可添加微信:mzbj0002 转账,或直接在文末打赏。

GEO数据下载
GSE70866有两个GPL,需要分别提取并注释。
rm(list = ls())
BiocManager::install("GEOquery")
library(GEOquery)
eSet <- getGEO(GEO = 'GSE70866',
destdir = '.',
getGPL = F)
# 提取表达矩阵exp
exp1 <- exprs(eSet[[1]]) #GPL14550
exp2 <- exprs(eSet[[2]]) #GPL17077
exp1[1:4,1:4]
dim(exp1)
dim(exp2)
#exp = log2(exp+1)
# 提取芯片平台编号
gpl1 <- eSet[[1]]@annotation
gpl2 <- eSet[[2]]@annotation
gpl1
gpl2
## GPL注释
library(devtools)
install_github("jmzeng1314/idmap3")
## 下载后本地安装
## devtools::install_local("idmap3-master.zip")
library(idmap3)
ids_GPL14550=idmap3::get_pipe_IDs('GPL14550')
head(ids_GPL14550)
ids_GPL17077=idmap3::get_pipe_IDs('GPL17077')
head(ids_GPL17077)注释基因表达矩阵并合并。
library(dplyr)
exp1 <- data.frame(exp1)
exp1$probe_id = row.names(exp1)
exp1 <- exp1 %>%
inner_join(ids_GPL14550,by="probe_id") %>% ##合并探针信息
dplyr::select(-probe_id) %>% ##去掉多余信息
dplyr::select(symbol, everything()) %>% #重新排列
mutate(rowMean =rowMeans(.[grep("GSM", names(.))])) %>% #求出平均数
arrange(desc(rowMean)) %>% #把表达量的平均值按从大到小排序
distinct(symbol,.keep_all = T) %>% # 留下第一个symbol
dplyr::select(-rowMean) #去除rowMean这一列
exp2 <- data.frame(exp2)
exp2$probe_id = row.names(exp2)
exp2 <- exp2 %>%
inner_join(ids_GPL17077,by="probe_id") %>%
dplyr::select(-probe_id) %>%
dplyr::select(symbol, everything()) %>%
mutate(rowMean =rowMeans(.[grep("GSM", names(.))]))
arrange(desc(rowMean)) %>%
distinct(symbol,.keep_all = T) %>%
dplyr::select(-rowMean)
exp = exp1 %>%
inner_join(exp2,by="symbol") %>% ##合并探针信息
tibble::column_to_rownames(colnames(.)[1]) # 把第一列变成行名并删除
# 先保存一下
save(exp, eSet, file = "GSE70866.Rdata")
# load('GSE70866.Rdata')
# install.packages("devtools")
# 提取临床信息
pd1 <- pData(eSet[[1]])
pd2 <- pData(eSet[[2]])
## 筛选诊断为IPF的样本
pd1 = subset(pd1,characteristics_ch1.1 == 'diagnosis: IPF')
pd2 = subset(pd2,characteristics_ch1.1 == 'diagnosis: IPF')
exp_idp = exp[,c(pd1$geo_accession,pd2$geo_accession)]批次校正
## 批次校正
BiocManager::install("sva")
library('sva')
## 批次信息
batch = data.frame(sample = c(pd1$geo_accession,pd2$geo_accession),
batch = c(pd1$platform_id,pd2$platform_id))未批次校正前PCA
#install.packages('FactoMineR')
#install.packages('factoextra')
library("FactoMineR")
library("factoextra")
pca.plot = function(dat,col){
df.pca <- PCA(t(dat), graph = FALSE)
fviz_pca_ind(df.pca,
geom.ind = "point",
col.ind = col ,
addEllipses = TRUE,
legend.title = "Groups"
)
}
pca.plot(exp_idp,factor(batch$batch))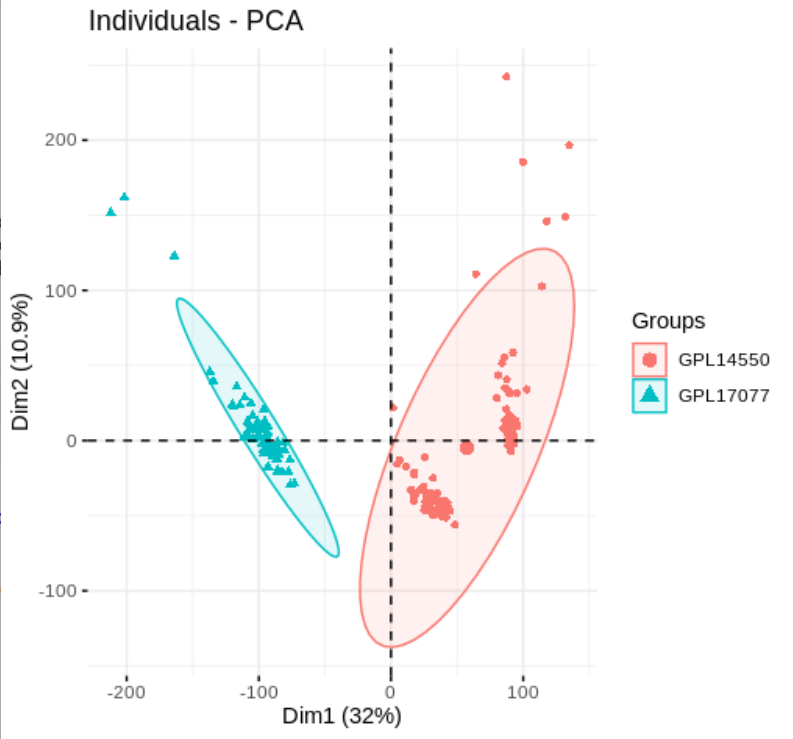
批次校正
## sva 批次校正
combat_exp <- ComBat(dat = as.matrix(log2(exp_idp+1)),
batch = batch$batch)
pca.plot(combat_exp,factor(batch$batch))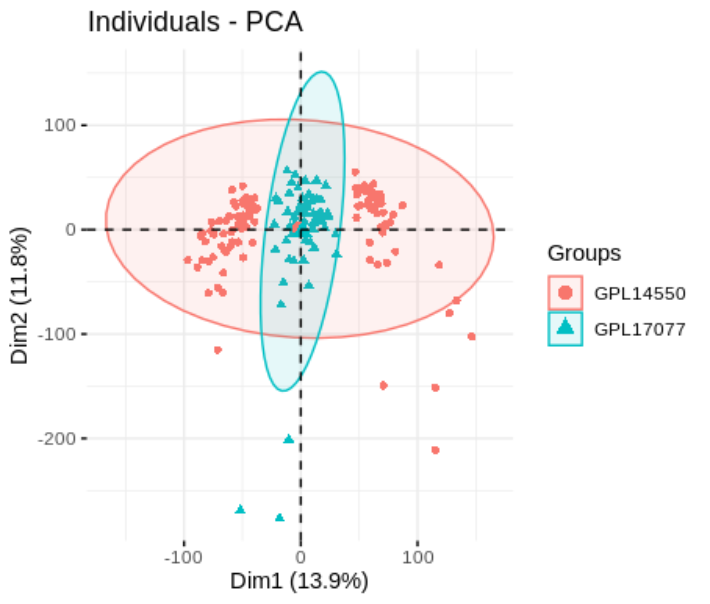
## 保存校正后的基因表达矩阵便于后续分析
save(combat_exp, eSet, file = "GSE70866.Rdata")注:因为只是浅复现一下,过程中可能会有些纰漏或瑕疵,希望大家批评指正。
往期内容
