WHISTLE特征提取
论文:

发表期刊:Nucleic Acids Research(JCR-Q1,11.5)
位点预测任务传统的特征:Sequence-derived feature
WHISTLE中加入的新的特征: Genome-derived features
WHISTLE集成35个Genome-derived features与传统Sequence-derived feature,重建了m6A表转录组的高精度地图
Genome-derived features:
基于序列特征本身并不能完全捕获RNA修饰拓扑结构的属性。各种研究已经证实了m6A RNA甲基化的生物学意义,包括在生物钟中的作用、调控mRNA翻译、热休克反应、microRNA (miRNA)处理、DNA损伤反应、RNA -蛋白相互作用和调控RNA稳定性。传统的位点预测方法只基于序列衍生信息(sequence-derived information)—即使使用了二级结构或其他高阶特征,信息仍然是直接从序列中提取的,而不考虑其他潜在的有用的基因特征。虽然序列信息可能起着核心作用,但是其他基因组特征可能也有助于预测m6A位点,因此我们产生了35个额外的基因组特征,可能有助于预测。
序列特征通过三种不同的结构化学性质:环结构、官能团和氢键进行编码核苷酸序
WHISTLE从多基因组特征中预测全转录组whole-transcriptome m6A位点,该框架基于多种基因组特征提取了一套完整的的领域知识,并将其与传统的序列衍生特征整合,进而高精度的预测m6A位点。然后,“内疚关联”guilt-by-association原则被应用于通过整合基因表达谱、RNA甲基化谱和PPI网络,进一步注释每个RNA methylation位点的功能相关性
WhistleR包使用
WhistleR: extract genomic features for predictive modeling in genomics
该包目前实现了两种类型的特征提取模块:基因组衍生特征和序列衍生特征。 前者是通过基因组属性与基因组区域相互作用获得的特征,后者包括one-hot编码的核苷酸序列(nucleotide compositions)或通过伪核苷酸组合(PesTNC)进行编码。
WhistleR安装:使用R devel版 R>=4.1,可以通过Github安装
可以将Granges格式的文件作为输入
1.1.1 Genome-derived features encoding(基因组衍生特征编码)
基因组衍生特征编码为基因组区域(例如,从基因注释中提取的区域:5'UTR, CDS, 3'UTR)与基因组间的特性(例如,x长度,GC含量)之间的相互作用,因此我们将这种特征编码方法命名为区域特性特征。
1.1.2 Sequence-derived feature encoding(序列衍生特征编码)
1.2 WhistleR安装

if(!requireNamespace("BiocManager",quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("WhistleR")
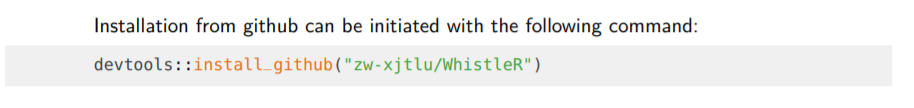
安装流程:
先安装R devel版 R>=4.1
下载:https://cran.r-project.org/bin/windows/base/rdevel.html
如果使用github安装还需安装devtools
通过install.packages("devtools")或者
if(!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("TxDb.Hsapiens.UCSC.hg19.knownGene")
安装
TxDb.Hsapiens.UCSC.hg19.knownGene
phastCons100way.UCSC.hg19
BSgenome.Hsapiens.UCSC.hg19
1.3工作流程
1.3.1从文件中读取基于范围的注释
如果目标注释存储在BED、GTF或GFF文件中,则可以使用rtracklayer包中的import()函数轻松地将其加载到R中。文件的格式可以直接从文件扩展名获得,也可以通过format参数手动指定。import()的输出是一个GRanges对象,可以直接作为特征提取函数的输入。
bed_dir <- system.file("extdata", "GSE63753.bed",
package = "WhistleR")
X <- rtracklayer::import(bed_dir)
当加载以其他表格格式保存的注释文件时,例如TXT或CSV文件,应该首先使用read.table()或read.csv()将表加载到R中。然后,可以使用在GenomicRanges包中定义的构造函数GRanges()从加载的data.frame中构造GRanges;该函数需要提供seqname(染色体信息)、注释的开始、结束/宽度和链的属性。
1.3.2 Genome-derived features基因组衍生特征
在WhistleR的标准工作流程中,我们将首先使用genomeDerivedFeatures()函数来提取基因组衍生的特征。一般来说,函数返回的特征矩阵采用1.1.1中描述的区域属性编码方法进行编码,并从函数参数中指定的标注对象中提取相应的区域和属性。本节的以下部分将解释基因组特征提取的具体用法和基本原理。
首先加载特征提取所需的包
library(WhistleR)
library(TxDb.Hsapiens.UCSC.hg19.knownGene) ##Txdb for transcript annotation
library(BSgenome.Hsapiens.UCSC.hg19) ##BSgenome for genome sequence
library(phastCons100way.UCSC.hg19) ##GScores for genomic scores
将注释包的变量更改为简洁的名称:
txdb_hg19 <- TxDb.Hsapiens.UCSC.hg19.knownGene
genome_hg19 <- BSgenome.Hsapiens.UCSC.hg19
phastCons_hg19 <- phastCons100way.UCSC.hg19
接下来,提取目标GRanges对象的基因组衍生特征。每个列的详细结构可以通过str()函数显示(在文档中不计算)。
RPF <- genomeDerivedFeatures(X,
## Providing gene annotation
transcriptdb=txdb_hg19,
## Providing genome sequence
sequence=genome_hg19,
## Providing Phastcons scores
gscores=phastCons_hg19,
## Calculate clustering metrics on X itself
clusteringY=X)
## Display the outcomes of feature extraction
str(RPF)
表1总结了从转录数据库指定的转录注释中提取的默认基因组区域

表1中列出的所有基因区域都是使用包基因组特征中定义的函数计算的。默认情况下,获得的启动子是转录起始位点(transcription start sites, TSS)的上游2000bp和下游200bp。