Article Directory
【2020-05】Application of next-generation sequencing in high-sensitivity detection of tumor mutations: challenges, progress and applications (review) 5.341 (Region 3 Q1)

Literature summary
The literature describes sources of error in the NGS detection workflow, as well as an introduction to non-NGS detection methods and their advantages and disadvantages. Specifically include:
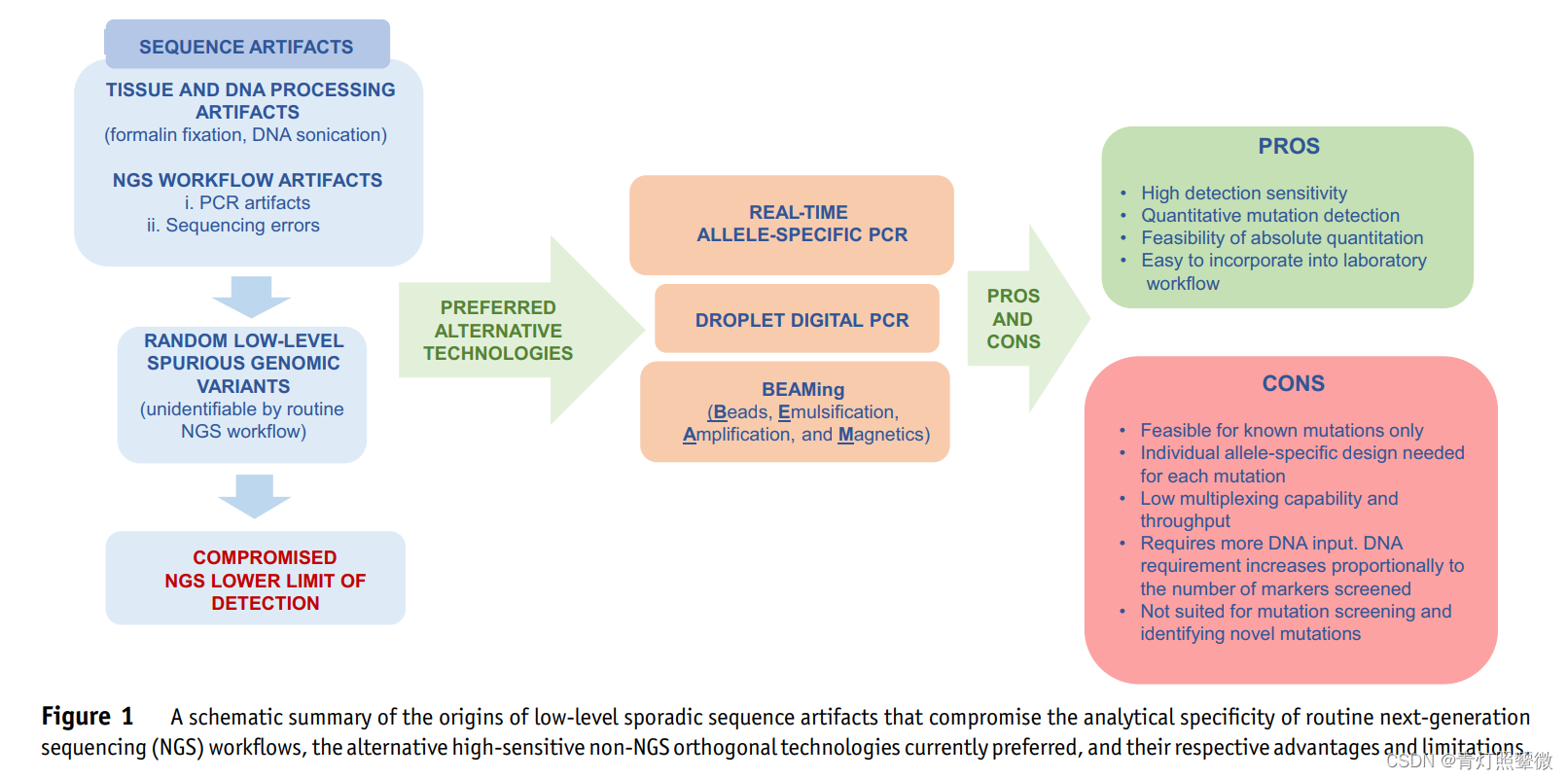
1) The source of sequence artifacts (artifacts, artifacts/errors/artificially processed)
- Tissue and DNA processing: FFPE/formalin fixation, DNA ultrasonic disruption
- NGS processing: PCR amplification, sequencing errors
- Random low-level pseudogenetic mutations (unrecognized by routine NGS workflows)
- Compromised NGS Lower Limit (LOD) (Lower LOD of NGS is limited)
2) Preferred alternative technique (non-NGS):
- Real-time multispecific PCR;
- ddPCR: droplet digital PCR;
- BEAMing: digital PCR flow cytometry (simple version of ddPCR);
BEAMing is an improvised version of ddPCR
3) Advantages and disadvantages of non-NGS
- advantage:
- High sensitivity;
- Quantitative mutation detection;
- Absolute quantitative feasibility;
- Easy to incorporate into laboratory workflow;
- shortcoming:
- Applies only to known mutations;
- Allele-specific design for each mutation;
- Low multiplexing capability and throughput (small amount of samples and data to accommodate?);
- More starting amount of DNA is related to the number of markers;
- Mutation screening and identification of new mutations are not applicable;
LOD(LIMIT OF DETECTION) is the detection limit, the LOD in the variation detection mutation frequency, generally, at the specified LOD frequency, 95% of the test samples will be reliably detected. For, finite gene sets, whole exome sequencing, and whole genome sequencing, 2%至15%it is not surprising that the reported LOD range is the variant allele frequency.
Sources of Error in NGS Workflows
1) FFPE sample:
Formalin fixation adversely affects nucleic acid integrity, extraction efficiency, and amplification: extensive DNA fragmentation, reduced DNA yield, chemical modification, C:G>T: A single-nucleotide artifacts, generation of abasic sites by depurination (generate non-basic sites by depurination). They result in reduced DNA amplification and induce artifacts, including single-nucleotide variants, short deletions, and chimeric and strand-split read artifacts. These artifacts are sporadic and nonspecific, often manifesting as low-level sequence variation.
These reduce errors by improving experimental conditions.
2) DNA interruption:
Produces low-level 8-oxoguanine lesions (low-level 8-oxoguanine lesions) leading to G:C>T:A artifacts.
3) PCR amplification and polymerase fidelity:
Both the amplicon method and target enrichment (probe capture) perform PCR amplification after the target sequence is obtained. The accuracy of sequencing depends on the quality/quantity of the DNA, the number of PCR cycles, and the fidelity of the polymerase and its ability to handle nucleic acid artifacts.
Because both of these methods rely on PCR, the final sequencing accuracy is dependent on the quality and quantity of DNA, the number of PCR cycles, and the fidelity of polymerases and their ability to deal with the nucleic acid artifacts.
Although high-fidelity polymerases have an error rate of one million base pairs (error rate 1 0 − 6 10^{−6}10− 6 ) Often used in NGS workflows, the problem is compounded by the use of excessive PCR cycles and low-quality DNA, as well as processing-induced chemical modifications, leading to sequencing artifacts. In addition, polymerase-induced PCR errors during the clonal amplification of DNA strands (to isolate and amplify each DNA library strand for sequencing) and the actual sequencing by synthesis also contribute to additional errors. High GC- and AT regions are under-amplified.
4) Sequencing platform:
The overall sequencing error rate for the Illumina platform ranges from 0.25% to 0.8%. Ion Torrnt has an indel sequencing error rate of 1.1% to 2%, while a substitution error is not significant (0.04% to 0.1%)
5) Data analysis:
Sequence alignments, where the presence of repetitive sequences and the occurrence of complex insertions and deletions can lead to misalignments that can adversely affect variant calling.
NGS Workflow Error Resolution Strategies
use UID
Template tagging模板标记(UID):Inability to identify artifacts that are pre-analytic or during the first PCR cycle of UID inclusion (jackpot mutations)
Duplex sequencing double chain UID;
Single-molecule sequencing;
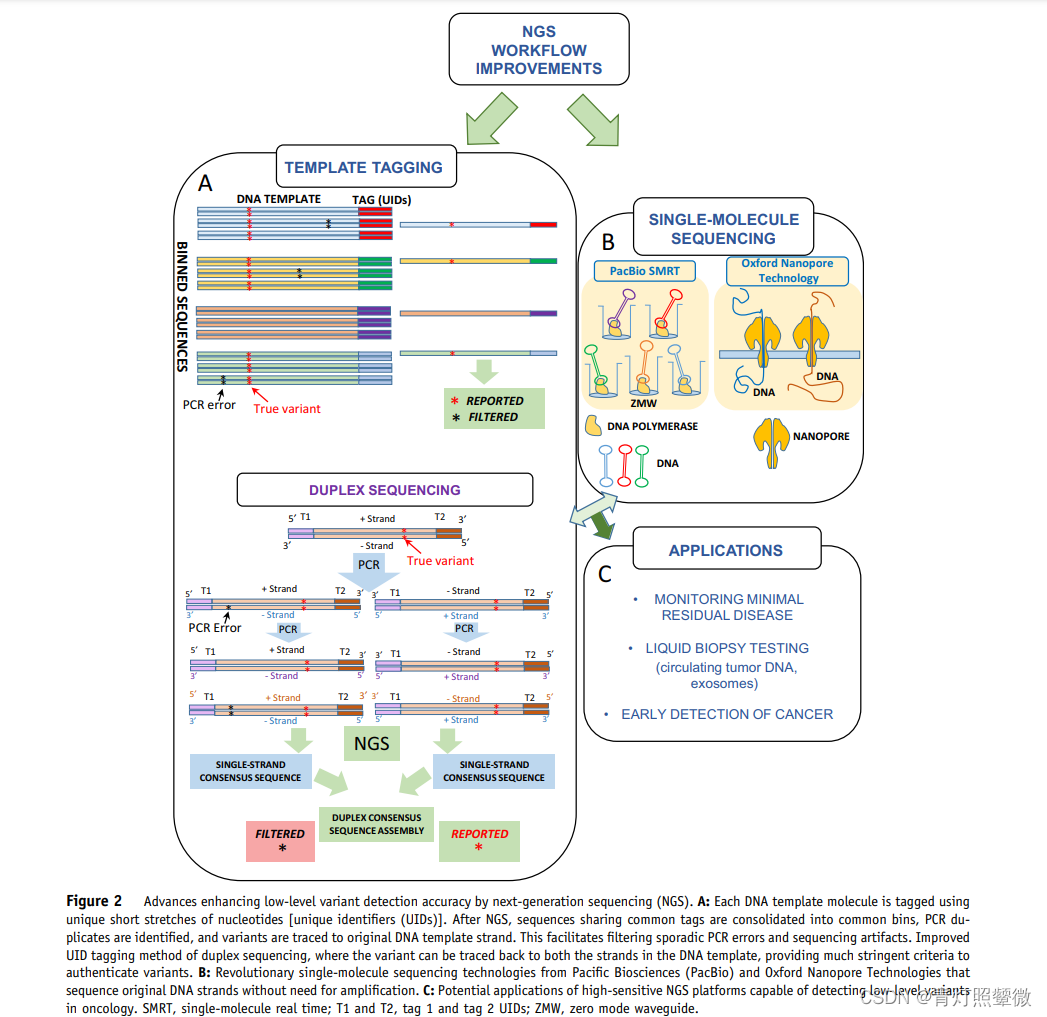
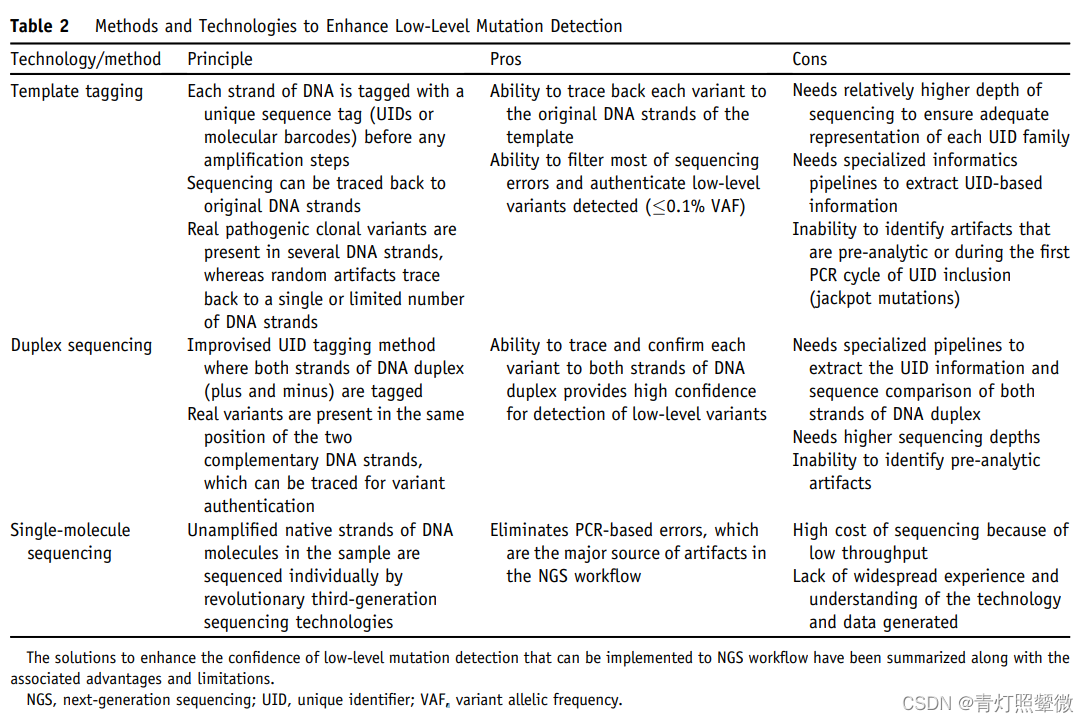
UID not used
Although some methods without UMI represent a major improvement over non-UID, applicability in routine clinical laboratories and performance to expected standards for clinical trials 尚未得到证明.
(1) Approach 1: Double-Strand Proximity Sequencing (Pro-Seq): A Method to Improve the Accuracy of DNA Sequencing Without the Redundant Cost of Molecular Barcodes 3.752 (Zone 3 Q2) 2018-10
Double-strand proximity sequencing physically links copies of each template DNA strand so that both double-strands are sequenced as a single cluster during the sequencing process, and mutants detected in a cluster can be traced back to both DNA strands (+ and − ), without using UID for double-strand sequencing
duplex proximity sequencing physically links copies of every template DNA strand so that both the duplex strands are sequenced as a single cluster during sequencing, and the mutant detected in the cluster can be* traced back to both DNA strands (+ and −)* without the need for duplex sequencing with UIDs
Pro-Seqis a novel approach to NGS library construction and analysis that achieves performance comparable to the best barcoding methods, but without the added cost of sequencing and subsequent sequencing. Multiple copies of each template are combined into a single sequence by physically linking copies of the molecule, thereby seeding a single sequencing cluster.
Using both cfDNA and cell line DNA, we report the average per-mutation detection threshold and per-base analytical specificity to be 0.003% and >99.9997% respectively.
(2) Approach 2: Barcode-free next-generation sequencing error verification for detection of ultra-rare variants 17.694 (Region 1 Q1) 2019-02
A cost-effective NGS error validation method is presented. By physically extracting and individually amplifying erroneously read DNA clones, we distinguished authentic variants with frequencies >0.003% from systematic NGS errors and selectively validated NGS errors after NGS. We achieved a PCR-induced error rate of 2.5 × 10-6 per base per doubling event, using a 10-fold reduction in sequencing reads compared to previous studies.
High-sensitivity detection on non-NGS platforms
1) Real-time fluorescent quantitative PCR:
The overall LOD was determined at 1 to 2 target molecules, and limits of quantitation were established at higher orders of magnitude (16 to 20 molecules). For variant detection, the LOD and limit of quantitation are as high as 0.0002%和0.0008%(2 to 8 variant strands established in a background of 1 million wild-type strands)
2) Droplet digital PCR (ddPCR):
The established 0.1%LOD for ddPCR (1000 variant copies on a background of 1 wild-type copy) can be improved by an order of magnitude or two if higher DNA input amounts are used ( ) 0.01%和0.001%.
3) BEAMing is an improvised version of ddPCR, the regular LOD of VAF is 0.01% 至 0.02%between , which can be improved to by some means <0.01%.
However, these platforms are adapted to detect known mutations, primarily one at a time.
NGS application
MRD:
-
MRD level LOD definition, which is usually
0.01%(one in ten thousand).(1) A recent study examined estimates of MRD in acute myeloid leukemia using a 24-gene NGS panel that adjusted for error suppression (limiting PCR errors by proofreading polymerases and low PCR cycle numbers) and error correction (molecular barcoding or UID). LOD up to
0.016%VAF.
Measurable residual disease monitoring by NGS before allogeneic hematopoietic cell transplantation in AML(2) Another NGS study used a panel of four genes (IDH1, IDH2, NPM1, and FLT3) with an error-correction approach that only tracked hotspot mutations and statistically filtered position-specific sequencing artifacts. LODs were established by flow cytometry and quantitative ASPCR
0.01%(SNV)和0.001%(插入缺失).
A novel deep targeted sequencing method for minimal residual disease monitoring in acute myeloid leukemia - PubMed
ctDNA early screening:
(1) Previous research without UID :
- Amplicon: Two-step amplification of target regions in six genes with
2%LOD of VAF and specificity of 97% (before improvement). Improvement: Enhanced library preparation efficiency and statistical analysis algorithm, LOD of 36 lung cancer-related genes is as low as0.25%VAF. - Hybridization Capture: Personalized analysis which showed a median LOD of
0.1%VAF with a specificity of 99%. - However, in the absence of any error-correcting methods, the confidence of these methods may be considered insufficient to routinely screen for
<0.1%mutations in VAF.
(2) Use UIDs :
Screening of 54 cancer-associated genes for SNVs, indels, and gene copy number variations and fusions showed a 0.1%consistent LOD for VAFs as well as significant analytical specificity (99.999%). A larger 73-gene cancer-associated gene panel was validated as a clinical trial of ctDNA. This test indicates a detection sensitivity of 0.02%至0.04%LOD.