 长读长宏基因组测序揭示高拷贝小质粒为动物粪便微生物组中高流行耐药基因关键载体
长读长宏基因组测序揭示高拷贝小质粒为动物粪便微生物组中高流行耐药基因关键载体
Long-read metagenomic sequencing reveals that high-copy small plasmids shape the highly prevalent antibiotic resistance genes in animal fecal microbiome
Article, 2023-06-02, Science of the Total Environment, [IF 10.75]
DOI:10.1016/j.scitotenv.2023.164585
原文链接:
https://www.sciencedirect.com/science/article/pii/S0048969723032060
第一作者:Kai Peng (彭凯);Yong-Xin Liu (刘永鑫)
通讯作者:Zhiqiang Wang (王志强);Ruichao Li (李瑞超)
合作作者:Pengcheng Du (杜鹏程);Yunzeng Zhang (张云增);Mianzhi Wang (王勉之)
主要单位:
扬州大学兽医学院 (Jiangsu Co-Innovation Center for Prevention and Control of Important Animal Infectious Diseases and Zoonoses, College of Veterinary Medicine, Yangzhou University, Yangzhou 225009, China)
扬州大学比较医学研究中心 (Institute of Comparative Medicine, Yangzhou University, Yangzhou 225009, China)
中国农业科学院深圳基因组研究所 (Shenzhen Branch, Guangdong Laboratory of Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen 518120, China)
齐碳科技有限公司 (Qitan Technology, Chengdu 610042, China)
扬州大学农业科技发展研究院 (Joint International Research Laboratory of Agriculture and Agri-Product Safety, The Ministry of Education of China, Yangzhou University, Yangzhou 225009, China)
- 摘要 -
动物源性抗生素耐药基因的出现和流行对全球公共卫生构成了巨大威胁。长读长宏基因组测序越来越多地被用于破译环境中耐药基因的命运。然而,利用长读长宏基因组测序对动物源耐药基因的流行分布、共存模式以及宿主信息的研究很少受到关注。为探究动物粪便中耐药基因的赋存状态,作者采用了新兴的齐碳纳米孔长读宏基因组测序方法,对蛋鸡粪便中的微生物群落和抗生素耐药谱进行了全面系统的调查,并分析了耐药基因的宿主信息和遗传结构。结果显示,不同生长阶段的蛋鸡粪便中均存在高丰度且高度多样性的耐药基因,表明经济饲养动物的粪便是耐药基因富集以及维持的重要场所。耐药基因遗传位置分析显示,与质粒介导的耐药基因相比,染色体上的耐药基因分布模式与粪便微生物群落关联性更强。基于长读长数据的耐药基因宿主追溯结果则表明变形菌门中的耐药基因通常由质粒携带,厚壁菌门中的耐药基因多由染色体携带。共现分析表明,不同耐药基因的共选择现象在蛋鸡粪便微生物组中普遍存在,高度活跃的插入序列会导致多种耐药基因的严重流行。值得注意的是,长读长无偏倚宏基因组测序发现高拷贝小质粒在floR和tet(L)等高流行耐药基因的传播中发挥了重要作用,甚至会扰乱粪便中耐药基因的原始组成。综上所述,作者的研究结果扩展了对饲养动物粪便抗性组的全面认识,其对蛋鸡源多重耐药细菌的预防和管理具有重要意义。
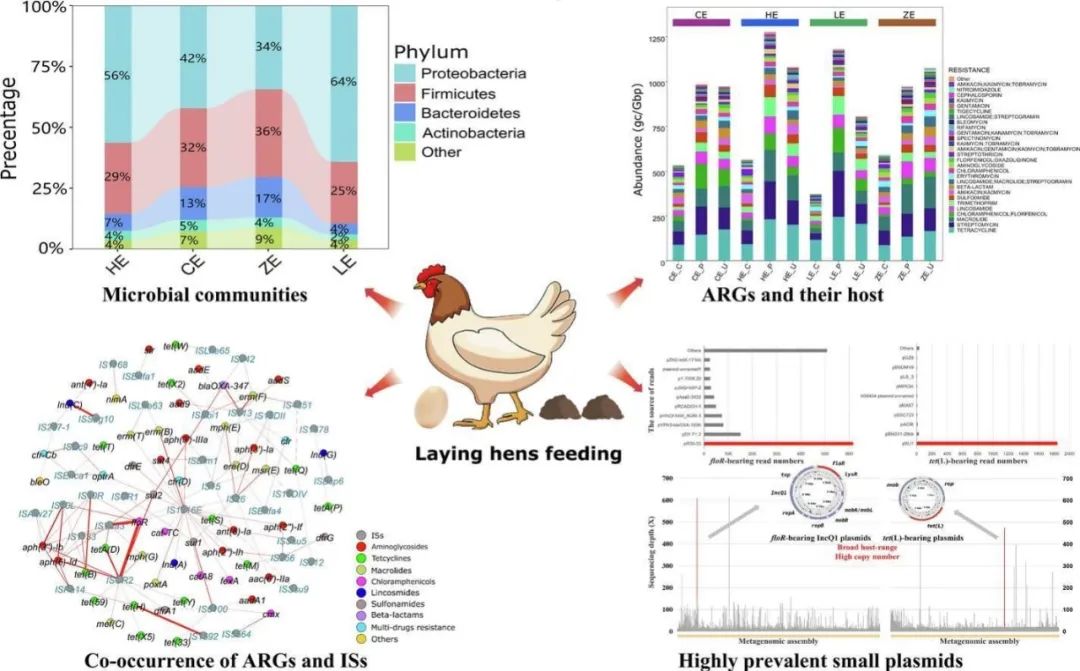
图形摘要
- 引言 -
抗生素已被应用于临床治疗及动物产业很多年,由于抗生素的滥用及过度使用而造成的耐药基因的出现和传播已经成为全球关注的公共卫生问题。过去,抗生素除了被用于细菌感染治疗以外,也被允许用于动物饲养过程中的饲料添加剂,导致了动物源耐药基因的严重流行,并且许多临床重要的耐药基因,如mcr-1,tet(X)和tmexCD-toprJ等,首次在动物源细菌中被发现。值得注意的是,已有研究确定动物源抗生素的使用会导致人类抗生素应用危机。在最近的几十年中,大约有75%的人类疾病是直接或间接起源于动物。因此,动物源多重耐药菌的出现和传播间接对人类细菌感染性疾病的治疗带来了很大的挑战。为控制耐药菌及耐药基因的传播,对动物源耐药基因的流行监测是必要且重要的。
宏基因组测序技术是一种不依赖于分离培养微生物就能获取微生物群落中遗传信息的新兴技术。其已被广泛应用于鉴定新的生物催化剂、酶和耐药基因,提出新的微生物功能假设以及快速检测细菌和病毒病原体。此外,宏基因组测序技术也也被用于监测肠道菌群以及环境菌群中耐药基因的流行。虽然传统的短读宏基因组测序在估计微生物群落中耐药基因的丰度方面表现良好,但难以对耐药基因遗传位置和宿主物种进行确定。相比较而言,长读宏基因组测序能够直接获取耐药基因的丰度、耐药基因宿主物种以及耐药基因的遗传环境等信息,使我们能对微生物群落中的耐药基因有一个更加综合、直观的了解。此外,长短读宏基因组测序的结合为耐药基因分析提供了更先进的分析方法,其能在菌株水平鉴定耐药基因的宿主信息。基于以上优点,长读长宏基因组测序是追踪复杂生态位中耐药基因命运的高效手段。当前,长读长宏基因组测序主要由牛津纳米孔公司测序公司以及太平洋生物科学公司提供。中国自主研发的齐碳纳米孔测序技术已被证明可高效获取多重耐药菌基因组完成图,但是其还未被应用于长读宏基因组测序领域,基于国产齐碳长读测序技术的宏基因组及耐药组研究值得探索。
鸡蛋是世界范围内营养丰富、价格实惠的食物,几乎所有的鸡蛋都是产自于大型蛋鸡养殖场,并且,蛋鸡相对肉鸡具有更长的饲养周期。因此,大型蛋鸡养殖场可能成为耐药基因富集的重要场所。之前已有研究通过短读宏基因组测序的方法研究鸡肠道微生物组中耐药基因的丰度及多样性,同时也有研究通过短读宏基因组组装的方式去探索蛋鸡源耐药基因的遗传位置及可移动性。然而,蛋鸡粪便中高流行耐药基因的遗传背景尚未通过长读长宏基因组测序得到很好的研究。本研究中,作者采用了新兴的国产齐碳纳米孔长读宏基因组测序技术,对蛋鸡粪便中微生物群落和抗生素耐药基因谱进行了全面系统的研究,同时分析了蛋鸡粪便中耐药基因的宿主信息和遗传结构特征(图1a),以上数据将对蛋鸡养殖过程中细菌中产生及富集的耐药基因的防控提供重要数据基础。
- 结果 -
1. 齐碳纳米孔宏基因组测序的基本序列特征
Basic features of the metagenomic sequences
从4个规模蛋鸡养殖场(分别为CE、HE、LE、ZE)的20个粪便宏基因组样本中测序产生44.9 GB的长读宏基因组数据,平均每个样本产生2.24 GB数据。齐碳纳米孔宏基因组原始数据的平均质量值为8.9,平均读长为1605 bp,读长N50为3340 bp。每个养殖场获取的宏基因组数据的读长N50从2848 bp到4350 bp不等,各养殖场中获取的宏基因组数据的平均读长均分布在1000 bp左右(图1bc)。
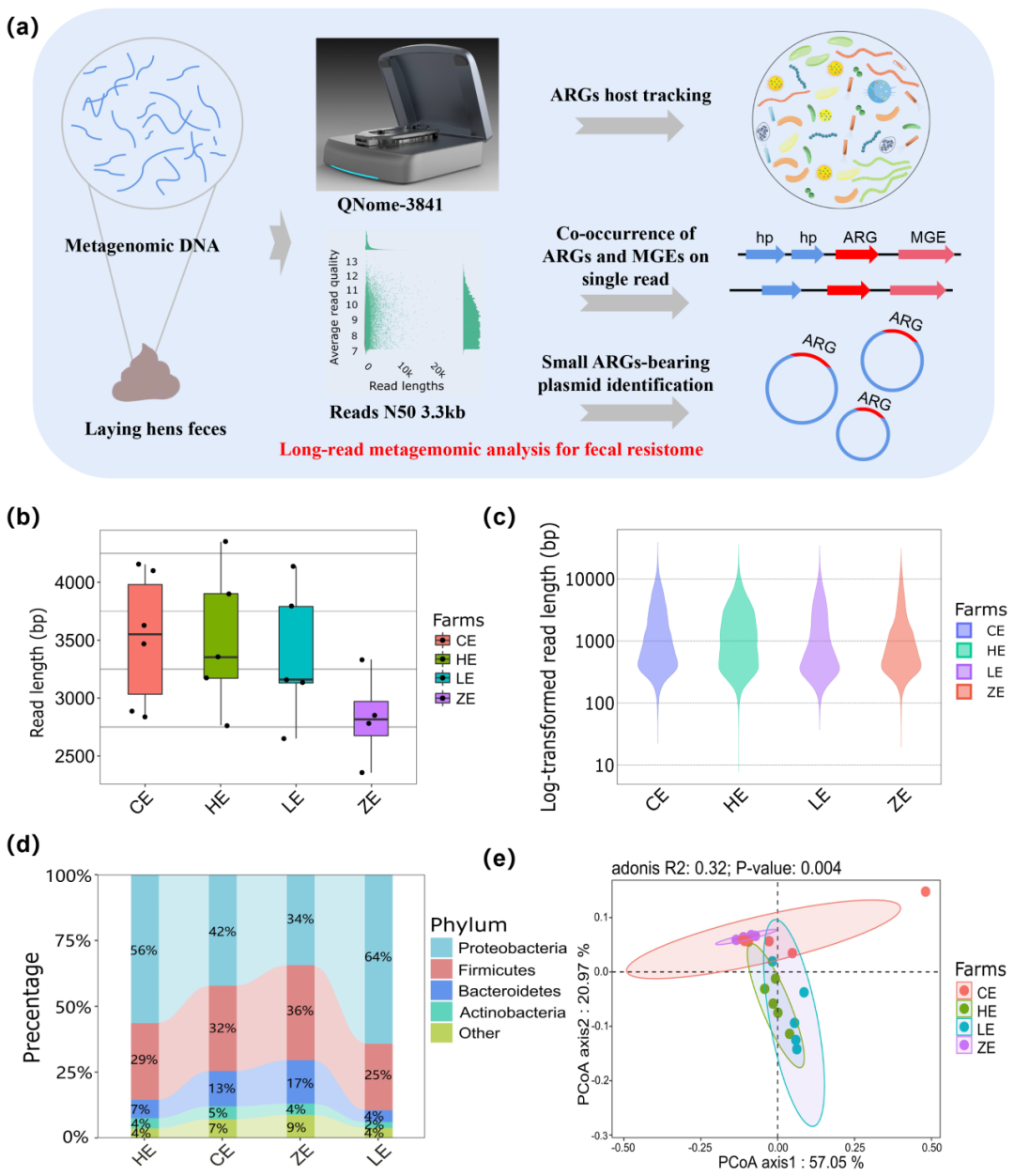
图1 实验设计,宏基因组序列特征以及蛋鸡微生物群落多样性。
(a)本研究的工作流程。首先提取宏基因组DNA,利用QitanTech纳米孔平台测序,最后进行生物信息学分析; (b)&(c)齐碳宏基因组测序读长N50分布及读长分布; (d)蛋鸡粪便中门水平的微生物物种组成; (e)属水平物种组成的PCoA结果。
2. 大型蛋鸡养殖场中蛋鸡粪便微生物群落结构特征
Fecal microbial communities of laying hens in large-scale farms
根据物种分类结果,作者发现不同养殖场的蛋鸡粪便微生物组成有明显差异。所有蛋鸡粪便样品中共鉴定出44个不同的细菌门,大多数蛋鸡粪便微生物群落由变形菌门、厚壁菌门和拟杆菌门细菌组成(图1d)。变型菌门的占比在HE和LE养殖场中显著高于CE和ZE养殖场,而拟杆菌门的丰度比例在CE和ZE养殖场高于HE和LE养殖场。LE和HE鸡场的蛋鸡分别处于停产期和育成期,CE和ZE鸡场的蛋鸡处于产蛋高峰期。两个处于产蛋高峰期的蛋鸡养殖场中粪便微生物组成相似,但是显著区别于停产期和育成期。随后,作者分析了蛋鸡粪便微生物组中丰度较高的属及种,丰度前5位的菌属分别是:Oceanisphaera(属于变形菌门)、Lactobacillus(属于厚壁菌门)、Pseudomonas(属于变形菌门)、Pseudoalteromonas(属于变形菌门)和Bacteroides(属于拟杆菌门)。LE养殖场中Oceanisphaera丰度显著高于其他3个养殖场。Oceanisphaera profunda,Pseudomonas sp. C27 (2019),Oceanisphaera avium,Pseudoalteromonas nigrifaciens和Pseudoalteromonas haloplanktis为丰度最高的五个种。随后的PCoA结果表明,不同生长阶段的蛋鸡粪便微生物组成在属及种水平均差异显著,与门水平的微生物组分布模式一致(图1e)。因此,蛋鸡粪便的微生物群落组成与蛋鸡生长阶段密切相关。
3. 蛋鸡粪便中耐药组特征
The resistome of laying hens fece
从整个数据集中共鉴定出462个不同的耐药基因,其中,193个耐药基因在四个养殖场中同时存在。不同样品中耐药基因的总丰度均处在较高的水平,波动范围为3085到5202个基因每GB数据。其中丰度最高的耐药基因为floR,tet(L),aph(6)-Id,aph(3'')-Ib,erm(B),sul2,erm(F),aph(3')-IIIa,aadA1和tet(M) (图2a)。floR和tet(L)基因在不同样品中的丰度差异显著。不同养殖场中耐药基因的分布模式无法通过丰度前50的耐药基因进行有效区分,因此,作者对不同养殖场中所有耐药基因进行了PCoA分析,结果表明,处于产蛋高峰期的CE和ZE养殖场中蛋鸡粪便中耐药基因组成模式相似,且明显区分于另外两个养殖场(图2b)。该结果与蛋鸡粪便微生物群落组成模式类似,因此推测蛋鸡粪便中耐药基因的存在模式受到微生物群落的影响。同时,普鲁克分析证实不论在属还是在种水平,蛋鸡粪便微生物组成均与粪便耐药组显著相关(图2c)。
为探究耐药基因的遗传位置,作者提取了所有携带耐药基因的序列进行了分类分析。应用Plasflow,可将携带耐药基因的序列分为质粒相关序列、染色体序列和未分类序列。经过遗传定位分类,作者发现染色体携带的耐药基因和质粒相关的耐药基因存在显著差异。此外,LEfSe分析显示,aph(3’)-IIIa、sat4和ant(6)-Ia是明显的染色体介导的耐药基因,而aph(6)-Id、tet(M)和aph(3’)-Ib多由质粒携带。随后,作者详细分析了丰度前100的耐药基因的遗传位置。其中,aac(6′)-Ib、aph(6)-Id、catA8、dfrE、lun(A)、floR、vga(C)等通常位于质粒上,而aph(3′)-IIIa、catA14、sat4、tet(H)等通常位于染色体上(图2d),这与LEfSe分析结果一致。此外,质粒介导的耐药基因比染色体介导的耐药基因更加丰度多样化,表明质粒在耐药基因的传播中起着更重要的作用。
Plasflow同时提供了序列的在门水平上的物种分类。结果发现,几乎一半的染色体序列和未分类序列属于未分类门。相比之下,大多数质粒相关的序列可以在门水平上有效分类。值得注意的是,几乎所有可分类的序列都属于变形菌门和厚壁菌门,这与粪便微生物组成一致。作者注意到部分携带耐药基因的染色体序列被归类为拟杆菌门。而在CE和ZE养殖场中,含有耐药基因的拟杆菌门的染色体序列所占比例大于在HE和LE养殖场。同时CE和ZE鸡场中蛋鸡粪便中拟杆菌门菌的比例也高于CE和ZE鸡场。这一发现进一步证明粪便微生物组成可能影响耐药基因的分布。然而,质粒相关的耐药基因受粪便微生物的影响较小,例如,LE和CE养殖场中染色体携带的耐药基因的序列占比有明显差异,但质粒携带的耐药基因的序列丰度没有明显差异。有趣的是,大多数携带耐药基因的质粒相关序列被分类为变型菌门,表明质粒通常出现在变型菌门细菌中。
随后,根据耐药基因的耐药表型,作者研究了不同样品中耐药表型的丰度。4个养殖场中发现的四环素类耐药基因最多,其次是链霉素、大环内酯、氯霉素/氟苯尼考和林肯胺类耐药基因。作者分析了耐药表型在染色体和质粒上的分布。大部分耐药表型由质粒携带,最为明显的是,氯霉素/氟苯尼考耐药基因大多位于质粒上,这一现象与大多数floR(氯霉素/氟苯尼考耐药基因)在质粒上检测到一致。同时,作者观察到ZE养殖场粪便样品中floR的丰度相对较低,相应的,ZE养殖场中氯霉素/氟苯尼考耐药表型比例与其他养殖场相比较低。由此可见,蛋鸡粪便中氯霉素/氟苯尼考耐药基因以质粒介导的floR基因为主。
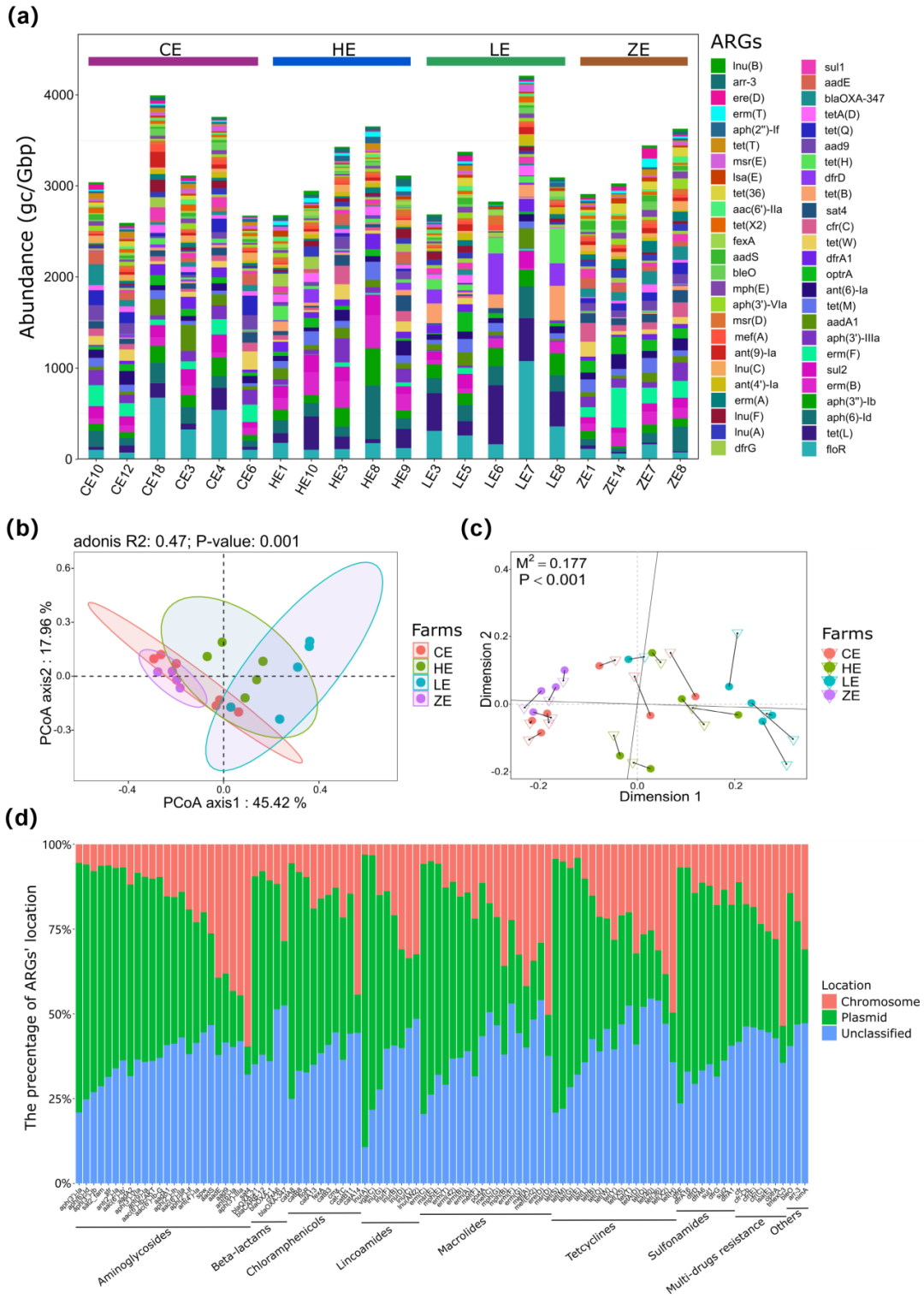
图2 蛋鸡粪便中耐药基因多样性。
(a)丰度前50的耐药基因在蛋鸡粪便微生物组中的分布; (b)不同养殖场耐药基因分布的PCoA结果; (c)耐药基因与物种组成关联的普鲁克分析; (d)蛋鸡粪便中丰度前100的耐药基因的遗传位置。
4. 不同蛋鸡养殖场中特有的细菌及耐药基因
The specific bacteria and ARGs in different laying hen farms
4个蛋鸡场中的微生物群落和耐药基因组成具有差异。为了鉴定4个养殖场中存在显著差异的细菌菌种和耐药基因,作者进行了LEfSe分析。ZE养殖场有16个LDA得分大于3的特有菌属,而其他3个养殖场分别有8个特有的菌属。CE、HE、LE和ZE养殖场中,链霉菌属、假互生单胞菌属、海洋菌属和拟杆菌属分别是富集最显著的菌属。此外,我们发现不同养殖场的耐药基因富集量与相应养殖场的菌属富集量接近,其中ZE养殖场中的特殊耐药基因最多。显然,更多样化的微生物群落通常会产生更多样化的耐药基因。tet(Q)、erm(B)、tet(L)和erm(F)分别是CE、HE、LE和ZE养殖场中最具代表性的耐药基因。此外,作者发现代表性的耐药基因通常由质粒携带,因此,一些携带耐药基因的质粒的流行和传播可能会扰乱耐药基因在粪便微生物群落中的流行模式。
5. 不同耐药基因之间及耐药基因与插入序列之间的共现模式
Co-occurrence of different ARGs and ARG with IS
携带耐药基因的长读序列的平均读长为3914 bp,足以编码一个以上的开放阅读框。因此,大多数携带耐药基因的序列都可进行基因的共现分析。根据结果,许多耐药基因被发现与其他耐药基因或插入序列共同位于单条序列上。aph(3”)-Ib和aph(6)-Id的共存在4个养殖场中最为常见(图3a)。sat4常与aph(3′)-IIIa和ant(6)-Ia共现,且频率较高。耐药基因遗传位置分析发现,aph(3’)-Ib和aph(6)-Id常见于变形菌门的质粒上,而sat4、aph(3’)-IIIa和ant(6)-Ia常见于厚壁菌门的染色体上。因此,耐药基因的共同进化发生在不同细菌的染色体和质粒上。值得注意的是,共现的耐药基因在微生物群落中的丰度是相似的,并且通常保持在较高水平。此外,对同一类抗生素具有耐药性的耐药基因通常同时发生,如aph(3”)-Ib-aph(6)-Id, sat4-ant(6)-Ia和tet(L)-tet(M)(图3a),该现象可能是由于抗生素的长期共选择造成的。同时,我们发现对不同抗生素产生耐药性的不同耐药基因也可能是共现的,如erm(F)-blaOXA-347、floR-aph(3”)-Ib和optrA-erm(A)(图3a),这类共存模式很可能导致细菌基因组中多重耐药区域的形成。
总体而言,4个养殖场中耐药基因的共现模式相似。值得注意的是,tet(H)基因的丰度在LE和HE农场比CE和ZE农场更高。作者进一步详细分析表明,高丰度的tet(H)与IS1592密切相关,特别是在LE养殖场的样品中(HE: 167/365;LE:592/998;ZE:2/44;CE:0/250)。此外,遗传定位分析表明,tet(H)通常位于变形菌门的染色体上。作者推测IS1592-tet(H)可能在染色体之间水平移动,导致tet(H)的严重流行。因此,共现分析可以直接监测微生物群落中耐药基因的活性状态。
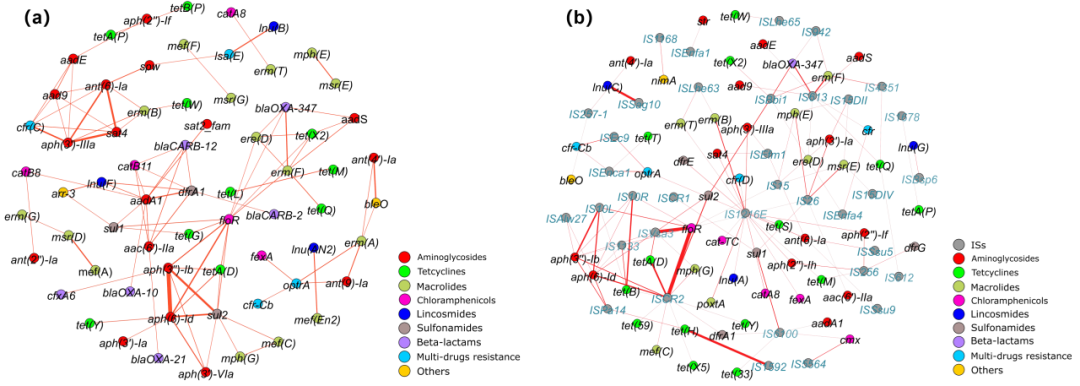
图3 不同耐药基因之间或耐药基因与插入序列之间的共现分析。
(a)不同耐药基因之间的共现分布模式; (b)不同耐药基因与插入序列之间的共现分布模式。
6. 高流行耐药基因的遗传背景分析
Genetic basis of highly prevalent ARGs
耐药基因的水平转移会加速它们的传播。耐药基因丰度分析表明,蛋鸡粪便微生物群落中含有丰富的floR和tet(L)基因。为了研究floR和tet(L)的高流行机制,作者提取了所有携带floR和tet(L)的序列进行了单分子遗传结构分析。为获得准确的结果,作者保留超过2000 bp的携带耐药基因的序列进行进一步分析。最终共5040条携带floR的序列作为查询序列,使用Blastn在质粒序列数据库中搜索相似的序列。其中,1621(1621/5040,32.16%)条序列与质粒数据库中某些质粒的序列总覆盖率超过85%。值得注意的是,548条(548/1621,33.8%)携带floR的序列与质粒pR39-33高度相似(图4a)。进一步分析发现,质粒pR39-33是由一个编码9个开放阅读框的8 kb的 IncQ型质粒的4个串联重复序列形成的。另外,69条携带floR的序列与质粒pZN5-floR-8kb具有较高的相似性。质粒pZN5-floR-8kb也是一个IncQ质粒,恰好为pR39-33的一个串联重复(图4a)。以上结果表明,蛋鸡粪便中大量携带floR的序列来自pZN5-floR-8kb及其衍生质粒。值得注意的是,我们通过共现分析发现ISCR2和ISVsa3在floR的传播中均起重要作用(图3b)。而在携带floR的IncQ小质粒中只检测到ISVsa3,说明ISCR2-floR对floR的传播也有很大贡献。
对于携带tet(L)的序列,与数据库中某些质粒总体覆盖率超过85%的序列比例(1980/4526,43.75%)显著高于携带floR的序列。有趣的是,它们中的大多数与质粒pSU1、pLS55和pBSDMV46A(1739/1980,87.8%)具有高度相似性,这些质粒为一组大小为5031 bp的相似质粒(图4b)。结构分析发现,这类质粒编码了可能参与质粒复制和移动的开放阅读框,表明它们是一类可转移质粒。此外,作者观察到高tet(L)丰度的样品中厚壁菌门的比例较低(HE和LE养殖场),但是,HE和LE养殖场中携带耐药基因的归类为厚壁菌门的质粒相关的序列比CE和ZE养殖场中更丰富。同时,遗传位置分析表明tet(L)主要由厚壁菌门的质粒携带。这一现象表明,携带tet(L)的小质粒的流行是HE和LE养殖场中厚壁菌门中tet(L)高流行的关键原因。
随后,作者分析了携带floR和tet(L)的序列读长分布,发现大多数携带floR的序列长度在8 kb左右,大多数携带tet(L)的序列长度在5 kb左右,这与携带floR和tet(L)的小质粒的大小相对应(图4cd)。这表明纳米孔长读宏基因组测序可以直接获取完整的小质粒序列。先前的研究表明,许多小质粒可以在细菌中形成串联重复结构,从而产生多种大小的质粒。我们发现,许多属于小质粒的序列的长度比小质粒的全长长。因此,在宏基因组数据中,两个小质粒的部分拷贝可能是以整个质粒的串联重复结构存在。然后,我们进行了单样本宏基因组组装。在CE3样品中发现了携带floR的质粒串联重复现象,在CE3、CE18和LE6样品中发现了携带tet(L)的质粒串联重复现象。同时,在组装结果中,我们也发现不同样品中携带floR和tet(L)的小质粒的测序深度不同。floR和tet(L)高流行率的样本中,携带floR和tet(L)的小质粒的测序深度也相对较高,说明小质粒在某些耐药基因的高度流行中起重要作用。除了携带floR和tet(L)的质粒外,我们还在长读宏基因组组装中发现了许多其他携带耐药基因的小质粒,这些小质粒携带的耐药基因包括lnu(A)、ant(9)-Ia、dfrD、optrA、cfr等。虽然这些小质粒在我们的数据中没有引起耐药基因的严重流行,但对这些质粒进行必要的关注对于耐药基因的监测非常重要。

图4 携带floR、tet(L)基因的小质粒分析。
(a)携带floR基因的序列与质粒数据库的比对结果; (b)携带tet(L)基因的序列与质粒数据库的比对结果; (c)&(d)宏基因组中含floR及tet(L)基因的小质粒的读长分布。
- 讨论 -
长读宏基因组测序在研究耐药组和微生物组方面具有许多优势。它可以高效的预测宿主物种信息和携带耐药基因的序列的遗传结构,使我们能够追踪耐药基因在不同环境生态位中的传播。齐碳纳米孔测序是出现在中国的一项新兴测序技术,其在细菌基因组测序中可以产生平均长度为6 kb的读长。本研究首次采用齐碳纳米孔宏基因组测序技术对蛋鸡粪便中的微生物组和抗性组进行了研究。研究基于连接法的文库制备方法,利用QNome-3848对蛋鸡粪便宏基因组进行测序,最终获取的蛋鸡粪便宏基因组序列的读长N50在2.8 kb ~ 4.2 kb之间,与之前使用牛津纳米孔长读宏基因组测序的研究相当。表明齐碳纳米孔测序产生的宏基因组数据读长适用于物种分类和质粒序列分类等分析。
作者在蛋鸡粪便中鉴定出44个不同门的细菌,以变形菌门、厚壁菌门和拟杆菌门为主,这与之前的研究结果一致。然而,变形菌门、厚壁菌门和拟杆菌门的丰度比例在不同的研究中差异很大,推测可能与物种分类方法和数据库差异有关。在当前研究中,作者使用centrifuge对微生物组成进行进行,并用分类至每种细菌的序列数量进行量化。不同鸡场的蛋鸡微生物群落组成表现出明显差异,但在同一生长时期的蛋鸡微生物群落表现出相似性。因此,蛋鸡粪便的微生物组成可能与蛋鸡的成长阶段关联更强。根据作者在采样前的调查,抗生素仅在蛋鸡育成阶段经常使用,然而,本研究在不同生长阶段的蛋鸡粪便中均发现了非常高的耐药基因丰度。作者推断,育成期中使用的抗生素可能会导致耐药基因的选择与富集,并持续蛋鸡一生,这与之前的研究结果表现一致。此外,作者发现耐药基因组成与属水平上的微生物群落的相关性大于种水平,这一现象表明,属于同一属的不同种类细菌可能会共享耐药基因,然而,更明确的耐药组和微生物组之间的关系值得进一步研究。
可移动元件对于耐药基因的传播至关重要。本研究结果表明,绝大多数携带耐药基因的序列属于质粒。值得注意的是,质粒介导的耐药基因在蛋鸡粪便中的比例与污水处理厂相当,这表明在许多环境中,质粒是耐药基因的关键储库。采用长读宏基因组测序的单分子分析方法分析耐药基因和插入序列的共发生,其结果比短读组装序列更全面、更准确。同时,基于原始单分子序列的共现分析可获取更丰富的数据集,可以生成更多样化的细菌基因组水平的共现模式。例如,作者在长读宏基因组数据中发现tet(X5)与ISCR2相关,虽然ISCR2-tet(X5)在宏基因组数据中很少受到关注,但它是细菌中介导替加环素耐药的重要遗传结构。因为在单菌基因组研究中,大多数授予替加环素耐药性的tet(X)变体是ISCR2动员。因此,基于长读序列的共现分析可用于预警一些重要耐药基因的早期流行。此外,我们通过长读单分子序列分析发现,许多高丰度的耐药基因通常与插入序列共存,这表明插入序列是蛋鸡粪便中耐药基因转移的重要遗传驱动因素。
作者的研究结果表明,小质粒可能是一些耐药基因在粪便微生物群落中高流行的原因。在部分蛋鸡粪便样本中检测到一种携带floR的IncQ型质粒和一种携带tet(L)的小质粒。这两个小质粒在粪便微生物中可以形成串联重复结构,进一步增强了耐药基因的富集。IncQ质粒是一种宿主范围广泛的质粒,其质粒骨架区域含有编码质粒复制和转移的模块。并且IncQ型质粒可以调动多种耐药基因,包括floR、tet(X)和blaCTX-M-3。此外,抗生素残留可能促进耐药基因和IncQ型质粒的富集。本研究仅在LE和CE农场的部分样品中检测到大量携带floR的IncQ型质粒,推测不同样品中的抗生素环境可能不同。此外,这种小的质粒可能会与包括其他质粒和转座子在内的广泛的遗传元件进行重组,使耐药基因更容易扩散。携带tet(L)的小质粒中也含有编码复制和移动的基因,表明它们也是高度活跃的。比较分析发现,本研究中发现的携带tet(L)的质粒与pMA67质粒几乎相同,pMA67质粒首次在蜜蜂的革兰氏阳性细菌病原体Paenibacillus幼虫中发现。随后,许多与pMA67相似的携带tet(L)的质粒在Lactobacillus、Bhargavaea、Sporosarcina和Bacillus中被鉴定出来,表明它们是一种宿主范围广泛的质粒。值得注意的是,携带tet(L)的小质粒组不仅介导了粪便中高度流行的tet(L),还可能导致tet(L)基因从鸡粪便传播到土壤中并持续存在。因此,这些小质粒的富集和转移对于耐药基因在不同生态位之间的传播至关重要。进一步研究携带耐药基因的小质粒的起源及其在不同环境中的长期命运至关重要。
参考文献
Kai Peng, Yong-Xin Liu, Xinran Sun, Qiaojun Wang, Pengcheng Du, Yunzeng Zhang, Mianzhi Wang, Zhiqiang Wang, Ruichao Li. (2023) Long-read metagenomic sequencing reveals that high-copy small plasmids shape the highly prevalent antibiotic resistance genes in animal fecal microbiome. Sci Total Environ, doi: 10.1016/j.scitotenv.2023.164585.
- 作者介绍 -
第一作者

扬州大学兽医学院
彭凯
博士研究生
扬州大学兽医学院博士研究生/中国农业科学院基因组所刘永鑫团队客座博士生,主要研究方向为细菌耐药性产生及传播机制、细菌基因组学、动物肠道宏基因组学等。目前以第一作者(或共一)在Science of The Total Environment,mSystems, Journal of Infections等期刊发表论文11篇。获国家奖学金、扬州大学科技先锋、扬州大学学术新人奖等奖励。

中国农科院基因组所
刘永鑫
研究员
刘永鑫,中国农科院基因组所研究员,iMeta期刊执行主编,宏基因组公众号创始人。主要研究方向为微生物组方法开发、功能挖掘和科学传播,在Science、iMeta、Nature Biotechnology、Nature Microbiology、Cell Host & Microbe、Protein & Cell等期刊发表论文50余篇,被引13000余次。主编《微生物组实验手册》专著,由300多位同行参与,共同打造本领域长期更新的中文百科全书。创办宏基因组公众号,15万+同行关注,累计阅读量超3千万,打造本领域最具影响的科学传播平台,免费为您团队发布成果、方法、经验、招生招聘等,欢迎投稿。2021年发起iMeta期刊,打造微生物组/生物信息领域国际顶刊,解决我本领域期刊出版卡脖子问题,建立国际学术话语权体系。
通讯作者

扬州大学兽医学院
王志强
教授、博士生导师
王志强,男,1972年4月生,中共党员,教授、博士生导师,现任扬州大学兽医学院院长。2000年毕业于华南农业大学,获兽医药理与毒理学博士学位,同年至扬州大学兽医学院任教。2009年-2011年在美国Auburn大学兽医学院进行访问学者研究。兼任中国兽药典委员、农业农村部兽药咨询专家、农业农村部全国饲料评审委员会委员、中国畜牧兽医学会兽医药理与毒理学分会常务理事、江苏省毒理学会副理事长、江苏省兽药协会副理事长、江苏省高校“青蓝工程”优秀青年骨干教师、江苏省“六大人才高峰”培养对象。主要从事畜禽重要病原菌耐药性传播机制及防控技术研究。先后主持国家自然科学基金面上项目、国家重点研发计划子课题等国家级、省部级科研项目30余项。曾获国家级教学成果奖二等奖、江苏省高等教育教学成果特等奖。近年来以第一作者或通讯作者在国内外学术期刊发表研究论文120余篇,其中SCI收录90余篇;主编或参编教材、专著10余部,授权专利4项。

扬州大学
李瑞超
教授,博士生导师
李瑞超,博士,扬州大学“青年百人”特聘教授,博士研究生导师,博士毕业于香港理工大学。目前主持包括国家自然科学基金面上项目等科研项目,入选人力资源与社会保障部2019年“高层次留学人才回国资助项目”(全国30个名额)人选。研究方向主要为兽医药理学和生物信息学,聚焦细菌耐药性和新型测序技术应用开发,掌握纳米孔单分子测序与分析技术,具体研究方向为:(1)细菌耐药性,运用微生物学、分子生物学和生物信息学技术,进行耐药性快速检测和耐药基因转移机制研究;(2)新型测序技术应用,运用高通量测序技术和单分子测序技术(如纳米孔测序技术等),进行细菌基因组学和耐药环境基因组学研究。近五年,已在本学科国际知名期刊(包括Engineering,GigaScience,Science of The Total Environment和Journal of Infections等)发表SCI论文60余篇,其中第一和通讯作者文章37篇(单篇最高引用151),一篇论文入选ESI高被引论文,总引用2500余次(Google Scholar);相关研究成果受到中国科学报等媒体报道。
猜你喜欢
iMeta简介 高引文章 高颜值绘图imageGP 网络分析iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读