对DNA深度测序数据进行去噪-高通量测序错误及其校正
抽象
表征常见的高通量测序平台所产生的错误并告诉技术伪像真正的遗传变异是两个相互依赖的步骤,这对于许多分析(例如单核苷酸变异调用,单倍型推断,序列装配和进化研究)至关重要。随机和系统错误都可以显示此处调查的六个主要测序平台各自的特定发生情况:454焦磷酸测序,完整基因组DNA纳米球测序,Illumina合成测序,离子洪流半导体测序,Pacific Biosciences单分子实时测序和牛津纳米孔测序。在对读取的数据进行排序时,有各种各样的程序可用于消除错误,这些程序在使用的错误模型和统计技术上有所不同,他们分析的数据的特征,他们从中确定的参数以及他们使用的数据结构和算法。我们重点介绍它们所做的假设以及这些假设所适用的数据类型,并提供指导以考虑使用哪些工具对数据属性进行基准测试。尽管此处未包含基准测试结果,但此类特定的基准测试将极大地指导工具选择和未来的软件开发。独立错误校正器以及单核苷酸变异体和单倍型调用者的开发,也可以从使用更多有关错误概况的知识以及(重新)组合此处介绍的现有方法中获得的思想中受益。我们重点介绍它们所做的假设以及这些假设所适用的数据类型,并提供指导以考虑使用哪些工具对数据属性进行基准测试。尽管此处未包含基准测试结果,但此类特定的基准测试将极大地指导工具选择和未来的软件开发。独立错误校正器以及单核苷酸变异体和单倍型调用者的开发,也可以从使用更多有关错误概况的知识以及(重新)组合此处介绍的现有方法中获得的思想中受益。我们重点介绍它们所做的假设以及这些假设所适用的数据类型,并提供指导以考虑使用哪些工具对数据属性进行基准测试。尽管此处未包含基准测试结果,但此类特定的基准测试将极大地指导工具选择和未来的软件开发。独立错误校正器以及单核苷酸变异体和单倍型调用者的开发,也可以从使用更多有关错误概况的知识以及(重新)组合此处介绍的现有方法中获得的思想中受益。
测序平台及其错误
我们首先调查五个常用的高通量测序平台在测序过程中产生的错误:454 [ 1 ]的GS FLX和GS Junior ,Illumina的Complete Genomics平台[ 2 ],HiSeq和MiSeq [ 3 ],个人基因组机(PGM)离子洪流[ 4,5 ]和实时测序仪(RS)由Pacific Biosciences公司[ 6 ]。此外,我们还简要介绍了Oxford Nanopore [ 7 ]最近发布的有关MinION平台的内容,但尚未向公众发布。有关基础技术和其他平台的详细评论,请参见其他地方[ 8,9 ]。对于除MinION之外的所有平台,都存在独立的错误评估,但是只有很少的研究系统地比较了多个平台[ 10–13]-覆盖范围不超过四个。同样,分析的重点也有所不同,仅报告一些众所周知的错误类型:插入和删除(通常归为插入/缺失),替换和覆盖率偏见,例如某些区域的覆盖率降低。为了确定是否在文库制备之前(例如,在预扩增步骤中),文库制备和扩增期间或测序运行中引入了错误,需要在不同实验条件下进行对比实验。这种时间和成本密集的分析很少进行,因此,仅在少数情况下做出了这种区分[ 13]。已知核酸序列的某些特性会提高所有或大多数技术的错误率,例如,GC含量的极限,较长的均聚物延伸,人类启动子序列的存在以及众所周知的碱基信号沿每次读取的衰减。在讨论了各个平台的错误情况之后,我们通过对所有平台在这些方面的直接比较来结束本综述的第一部分。
454焦磷酸测序
对于454个焦磷酸测序仪,已经报道了GS FLX [ 14 ]和GS Junior [ 12 ]机器的总体错误率,并评估了GS Junior [ 10 ]的插入缺失率。所有这三项研究仅研究了中等GC含量的序列。不过,他们报告的错误率(表1)支持众所周知的共识,即使用这项技术,插入错误比替换错误发生的频率高一个数量级。这种较高的插入缺失错误率主要是由于均聚物的出现,即同一核苷酸的多次连续出现。对于更长的均聚物长度,测序反应中各个基本流动循环的光强度分布越来越重叠,从而导致碱基调用中的插入和缺失错误[ 18 ]。由于这种现象,均聚物具有比其它序列段[更高的总插入缺失错误频率10,14 ],并用均聚物长度[在插入缺失错误频率的增加10。
高通量测序平台的错误率(每100个测序碱基)
| 平台 | 潜艇 | SD子 | 因德尔斯 | SD Indels | 一个所有 | SD全部 |
|---|---|---|---|---|---|---|
| 454 GS FLX | 0.09000 | b N / A | 0.90000 | b N / A | 0.99000 | b N / A |
| 454 GS青少年 | 0.05430 | b N / A | 0.39055 | b N / A | 0.45540 | b N / A |
| 完整基因组学 | 2.30000 | b N / A | 0.01900 | b N / A | 2.31900 | b N / A |
| Illumina HiSeq | 0.26400 | 0.11238 | 0.02561 | 0.02351 | 0.28467 | 0.11875 |
| Illumina MiSeq | 0.24551 | 0.11079 | 0.00905 | 0.01436 | 0.29652 | 0.18867 |
| 离子洪流PGM | 0.16985 | 0.17253 | c 1.45793 | c 1.21924 | c 1.63112 | c 1.24217 |
| 太平洋生物科学 | 1.10286 | 0.44761 | 15.56571 | 3.29386 | 16.19250 | 3.16667 |
仅根据足够大的样本量提供标准偏差的数字以粗体显示。
Subs =每100个碱基的替换错误;插入缺失=每100个碱基的插入和缺失错误;SD =标准偏差
a一些研究仅包含插入缺失和/或总误差的汇总量度。因此,All的值不一定是Subs和Indels之和。
b对于这些平台,只有一个样本可用。因此,由于没有SD,因此应谨慎考虑错误率。
c一项包含3个样本(总共12个样本)的研究使用了耐indel的作图,导致几乎100%的读数被作图,但产生的indel错误率也更高。此样本还说明了高SD。
454测序数据还包含相当数量的歧义碱基调用(某些调用者然后输出“ N”),尽管其频率远低于插入缺失的频率,并且与不匹配的频率相当[ 14 ]。向读出结束时,不明确的碱基调用频率显著增加,因为这样做替代错误,而错误插入缺失仅显示轻微但显着增加[ 14,18 ]。在读数的某个点之后,取决于所使用的机器和化学性质,GC含量(所有读数的平均值)也会急剧下降,这表明在以后的流量循环中,GC偏差很大([ 19 ]中的图1 )。同时,较长的读取令人惊讶地具有较低的平均错误率。Gilles等。[ 14 ]建议读取的错误率始终较低或始终较高,即较短的读取是高错误读取,已对它们进行了大幅度调整以消除最终的错误,但其余部分平均仍比较高的错误包含更多的错误。不需要过多修剪的高质量读取。最后,在该技术的微微孔板中发现了每孔插入和缺失的倒序模式:在测序板的某些部分中发现插入比发现缺失更常见,而在其他区域的效果却相反。因此,测序板的连续区域富集了缺失或插入区域,但这些模式在不同的测序板或运行之间似乎不一致(图3和[14 ])。

{kind=link}
对三种微生物(恶性疟原虫,大肠杆菌和球形红球菌)和人类基因组中不同局部GC含量的测序覆盖率。底部图显示了具有一定GC含量的各个基因组中100个碱基的窗口的相对分数。顶部面板显示了具有一定GC含量的100个碱基的窗口相对于各自平台样品平均值的相对测序范围。根据知识共享署名许可CC-BY 2.0(http://creativecommons.org/licenses/by/2.0/),此图是[ 13 ]中图2和图3的汇总和改编。该图的彩色版本可从BIB在线获得:http://bib.oxfordjournals.org。
完整的基因组DNA纳米球测序
完整基因组学DNA纳米球测序的错误信息来自人类基因组样品平台的系统比较[ 13 ]。考虑到这一限制,报告的错误率(表1)表明,与indel错误相比,该技术更常见的替代错误是两个数量级。发现总的错误率在很宽的GC序列内容范围内是一致的,除了具有较高或较低GC含量的序列的删除率要高得多(图2 B)。这两个GC含量的极端值还与较低的读取覆盖率相关(图1)。最后,插入缺失率随均聚物长度的增加而显着提高(图2)。一种; [ 13 ])。

{kind=link}
不同长度的均聚物中的错误率偏差,以及由于不同的局部GC序列含量而引起。(A)顶部面板显示每个基因组和平台长度不同的均聚物的平均错误率。(B)底部面板显示了100个碱基窗口的不同GC序列内容之间的错误率。根据知识共享署名许可CC-BY 2.0(http://creativecommons.org/licenses/by/2.0/),此图是[ 13 ]中图4和图5的汇总和改编。该图的彩色版本可从BIB在线获得:http://bib.oxfordjournals.org。

{kind=link}
概述如何根据纠错策略从读取集中生成堆积。(A)如果已知一个良好且紧密的参考,则可以将读取结果映射到该参考。否则,还需要其他方法之一:(B)读取的MSA可以由所有读取对的成对比对形成,所有读取在初始映射到可用参考的过程中都有重叠(灰色虚线从A到B) ),共享一个后缀的一部分的所有读取(从F到B的灰色虚线箭头)或共享一个k- mer种子的所有读取对中,由记录每个表均记录每个k- mer的所有读取的表标识(灰色表和相应的虚线的灰色箭头)。同样,所有k- mers计数的简单记录可用于推导(C)一个k- mer谱图或(D)一个汉明图(图4),并且将读后缀加上唯一符号($ x)的读后缀可用于构建(E)后缀特里(图5)或(F )的后缀数组(图6)。
通过合成进行Illumina测序
通过合成平台HiSeq和MiSeq当前Illumina测序的错误配置文件已被其特征在于大量的细节,对较早的Illumina平台,如1 G [严格分析还绘图20,21 ]和基因组分析仪II [ 22 ]。与替换错误相比,Indel错误的发生频率要低一个数量级,Illumina的总体错误率是所有技术中最低的(表1)。对于HiSeq,已显示在MiSeq数据中,缺失比插入[ 15 ]更常见,而插入比MiSeq数据[ 23 ]更常见。]。替换错误显示出某些替换的偏见:迄今为止,A C和G T转换最常见,在HiSeq数据中,每种转换约占全部替换的30%和25%[ 15 ]。对于MiSeq数据已经证明了类似的效果[ 23 ]。总体而言,朝向末端的误码率增大读取[ 15,23 ]时,可以通过修整低质量端被大多处理的问题,而且是在所述第二读取显著更高做在MiSeq配对末端测序[当23 ]。
在Illumina测序仪中,修饰的核苷酸会在流通池中循环冲洗。包含数百万个序列簇的流通池块(每个都进行单独的测序反应)排列在每个流通池的通道中。车道两端的磁贴趋于具有一些循环,平均错误率提高。这会导致空间聚集和流周期特定的错误[ 15 ],这种现象可能解释了使用相同测序库的不同测序运行之间错误模式的显着变化,因为相同模板不太可能位于边界在两个不同的运行中进行贴砖[ 13 ]。然而,在由相同材料制备的不同文库产生的序列之间可以观察到进一步的变异[ 13],说明在准备步骤中已经产生了错误。尤其是文库制备方法和测序引物已显示出引入错误偏倚[ 23 ]。同样,一些错误也与序列基序相关:尤其是长时间均聚物拉伸后(图2 A),富含GC的序列(图2 B; GGCGGG是最突出的基序)以及反向重复序列[ 11],插入缺失错误率增加。,13,22 ]。幸运的是,大多数这些错误率增加始终反映在各个读取位置的较低质量得分中[ 15]-然而,据报道,Illumina数据中普遍对基本通话质量进行了高估(例如 [ 21 ]中的补充图S1)和在文库或样品制备中引入的任何错误(例如,由于聚合酶链反应(PCR)扩增所致)都不会反映在测序质量评分中[ 23 ]。但是总的来说,这支持了对在读数[ 15 ]两端出现和积累的错误进行质量修整的实践,并呼吁在所有下游分析中进一步使用质量得分。此外,一些特定的序列错误股特有的,这也应该帮助他们辨别真正从一个多态性样本[在11, 15, 23]。最后,Illumina平台在极高和极低的GC序列含量下均显示出明显的读取覆盖率下降(图1),这是一种无需PCR的文库构建能够减少但不能完全消除的效果[ 13 ]。
离子激流半导体测序
对于Ion Torrent当前的半导体排序平台PGM,已经对错误进行了详细评估。在此,插入缺失错误的频率比替换错误高一个数量级,并且总错误率明显高于454和Illumina平台(表1)。如果以插入缺失耐受性定位读段,则对已知参考基因组进行测量时,插入缺失错误率甚至会更高:映射更多的读取,并计数更多的实际插入缺失[ 16 ]。研究之间的这种方法差异解释了表1中报告的错误率的高标准偏差。
均聚物[ 10 ]引起大量的插入缺失错误,碱基调用精度随均聚物长度[ 16 ]降低。一般而言,虽然在PGM数据中插入错误比删除错误更有可能发生,但整体而言,均聚物的删除错误更可能发生,并且均聚物的出现时间越长[ 16 ],导致均聚物长度的平均报告不足(图2 A)。 ; [ 13 ])。这对于超过约8个核苷酸的均聚物延伸尤为明显,并且可能最终导致超过14个核苷酸的均聚物的读取覆盖率完全丧失[ 11]。]。插入缺失错误的另一部分在参考基因组的某些位置以很高的频率发生[ 16 ]:这些错误大多是A或T插入或C或G缺失;其中约80%是特定于运行的(即需要测序重复以鉴定它们);其中约7%表现出明显的链特异性。这样的链特异性错误以前也有报道:一些连接到均聚物插入/缺失[ 10 ],另一些没有明显的序列基序连接[ 11 ]。
GC偏倚在PGM测序中具有明显的作用。尽管在极端GC序列内容中较高的总indel错误率比Illumina和Complete Genomics测序更稳定(图2 B),但序列覆盖率却没有(图1):在高和低GC序列含量下,其覆盖率都急剧下降在人类基因组中以及整个微生物基因组中具有不同的GC序列含量[ 13 ]。另有两项研究进一步证实了这一点,这些研究表明恶性疟原虫的极低GC基因组中存在很强的覆盖偏见[ 11 ],而马氏Deinococcus maricopensis的非常高GC含量基因组的文库制备完全失败。以及低硫含量的Sulfolobus tokodaii基因组的错误率升高[ 16 ]。因此,有人建议PGM文库的制备仅对(本地)GC序列含量约为20–80%的物种是安全的[ 16 ]。
最后,在朝向的端部平均增加错误率读取(图2 A; [ 10,16 ]),与错误率的一个明显的周期性,其连接到Samba流节律[ 16 ]:核苷酸被冲洗过的测序芯片分开处理,某些流(主要是A和T流)具有较高的呼叫率或呼叫率,分别直接与插入和删除错误相关。Samba流特定的错误模型可能允许更好的错误纠正[ 16 ]。
太平洋生物科学单分子实时测序
尽管报告的错误率(表1)比454焦磷酸测序和完整基因组学所支持的样本更多,但独立研究并未很好地描述Pacific Biosciences实时测序仪(PacBio RS)的错误特征,特别是关于PacBio的最新化学。较早的化学方法的总错误率比离子激流PGM的总错误率大大约一个数量级,比Illumina平台的总错误率大大约两个数量级(表1)。)。在平台内,插入缺失错误的普遍性是替代错误的15倍。错误率极高的缺点(以及较低的总吞吐量)使大型基因组(如人类基因组)[ 13 ]或元基因组研究中的DNA数量无法承受,其缺点被两个因素部分抵消:首先,读取时间长(〜10 kb)使该平台可用于较小基因组的从头序列装配的支架,该序列也使用来自另一个平台的读取数据[ 11 ]。其次,在极高的GC序列含量下,覆盖率仅略微下降,这使其成为具有最低GC偏倚的平台(图1; [ 13 ])。随着最近数据质量和读取长度的增加,仅从PacBio数据进行细菌基因组的从头组装就成为可能[ 24 ]。此外,平台供应商还提出了两种降低错误率的方法学方法:(i)SMRTbell模板是一种有效的圆形双链DNA模板,两端带有环,可以连续读取多个相同的模板。然后将它们汇总为具有较低错误率的共识读取[ 25 ]。(ii)或者,可以使用来自可变大小的不同模板的冗余覆盖范围来创建正确的共识。在这种方法中,越丰富的较短读取可提供覆盖范围的冗余,而较长的读取可确保装配的连续性[ 26]。这种额外的冗余通SMRTbell或增加的总覆盖-已经独立地示出通过一个数量级以减小总体误差率分别为1.3和2.5%,([ 27,28 ];部分“特定于平台的纠错”)。
此外,发现每次读取的最长部分的错误率是一致且随机的,较长读取的错误率仅稍有下降[ 17 ]。错误率也不受较长的均聚物拉伸的影响-缺失仅略微增加而插入的略微减少(图2 A)-以及在整个GC序列含量范围内(图2 B; [ 13 ])。
牛津纳米孔测序
像PacBio的实时测序一样,牛津纳米孔的MinION有望产生更长的读数,从而能够更好地解析结构变异和基因组重复内容。但是,这项技术比PacBio RS还要年轻,所有已发表的研究均基于平台开发人员的MinION Access Program [ 29 ]数据,并且仅基于DNA的短片段(噬菌体或细菌基因组或单个人类基因)。另外,化学正在快速发展,不断提高数据质量[ 30 ]。因此,应谨慎考虑此处提供的值,这些值既不包含在表1中,也不包含在下面的平台比较中。
与PacBio测序中的环状共有序列测序(CCS)读取类似,DNA发夹环连接到双链DNA分子的末端,将两条链连接在一起。但是,这仅在双链的一端完成,因此MinION仅对每个链进行一次测序[ 31 ]。这导致在发夹之前读取“模板”,并在发夹之后读取(反向)“互补”。只要有可能,就会建立两者的共识,即“二维”(2D)表示为[ 31 ]。大多数报告的错误率仅指这些二维读物,它们代表最新发布的流通池版本R7.3 [ 30–32 ]的约40%至50%的测序碱基,而其中只有63 [ 32 ]至90%[ 30]的[]映射到相应的参考。通过这样的阅读,Ammar等人。报告7%的替换错误,13.3%的删除错误和5.3%的插入错误。Jain等。进一步将其选择范围缩小到卖方碱基检出者标记为高质量的读物,最终分析了约24%的总测序核苷酸。平均所述噬菌体M13的超过三个重复,它们报告5.1%的取代的错误,7.8%缺失错误和4.9%插入错误,并且还应用他们的误差估计到大肠埃希氏菌快速的数据等。,他们发现5.3%替换错误,9.1%删除错误和6.0%插入错误。
总的来说,这些研究大致同意替换和插入错误的发生率相似,而删除错误的常见错误率约为两倍。联合错误率大约为20%到25%,仍然明显超过了其他当前的单分子测序技术PacBio。同样与PacBio相比,错误似乎有偏差:与取代错误相比,从A到T和从T到A的错误比所有其他取代错误[ 30 ]的可能性要小得多,并且均聚物运行似乎会增加插入和删除的错误率[ 30]。33 ]。
平台比较:与GC偏倚,均聚物和人类启动子序列有关的错误
在五个可用平台中的四个(不包括牛津纳米孔)上,已知具有极端GC含量的序列在产生的读数中覆盖范围减小,除了PacBio RS外,所有平台中的某些区域都没有读数覆盖(图1; [ 13,19 ])。对于使用GS FLX平台进行454焦磷酸测序,在一定的读取长度后,平均GC含量急剧下降是已知的GC偏差[ 19 ]。Ross等人系统地比较了其他四个平台的GC偏倚。[ 13 ]:所有平台均代表具有中等GC含量的序列,并且对高和低GC含量序列的覆盖率均降低(图1)。PacBio RS即使在极端的GC序列含量下也能提供最一致的覆盖范围,但目前尚不支持以合理的成本对大型基因组(例如人)进行测序。因此,在实践中,就微生物基因组分析而言,它在GC含量偏差方面仅胜过所有其他平台。离子激流PGM始终显示在微生物和人类基因组中,低和高GC序列含量区域的覆盖率损失最大。在低GC含量的基因组中,根本没有覆盖高达30%的基因组[ 11 ],而对于高GC含量的基因组,文库制备完全失败[ 16]。]。完整基因组学是专门针对人类基因组测序的平台,其性能类似于人类基因组上的PGM。在这里,Illumina的HiSeq平台的性能优于Ion Torrent PGM和Complete Genomics [ 13 ]。这些技术对极端GC内容序列的覆盖偏差也无法通过组合来自不同平台的数据来修正[ 13 ],这可能是因为偏差概况不是互补的,而是定性相似,只是GC偏差的强度不同。
此外,一些平台显示了GC含量或GC母题与不同种类的单核苷酸错误之间的相关性[ 13 ]。PGM的插入缺失率在整个GC范围内均保持稳定(总体水平较高),PacBio RS的插入缺失率甚至更高。完整的基因组学和Illumina具有较低的整体插入缺失率,但在极高和极低的GC含量下,缺失错误率均明显升高,Illumina数据中的插入误差也呈明显升高趋势(图2 B; [ 13])。Ion Torrent的PGM是唯一可以明显提高替代错误率的平台,尤其是在GC序列含量非常低的情况下,而Complete Genomics,Illumina HiSeq和MiSeq和PacBio RS在整个GC序列范围内的替代错误率都非常稳定内容,在极端情况下只有很小的升高(图2 B)。另外,Illumina数据中某些特定于链和特定于循环的错误已归因于富含GC的序列[ 11 ]。
随着均聚物的长度,所有平台示出了在插入错误率的增加,删除错误率或二者,除了PacBio RS与它一直很高INDEL错误率(图2 A; [ 13,14 ])。对均聚物最敏感的是Ion Torrent PGM,在一项研究中发现它对长于14个核苷酸的均聚物不产生任何读数[ 11 ]。
受轶事证据的启发,[ 13 ]还检查了人类启动子序列的覆盖范围,并确定了所有此类被测试平台(例如,Complete Genomics,Illumina HiSeq和Ion Torrent PGM)极度覆盖的此类启动子。既没有用GC序列含量也没有用均聚物解释。当与改良试剂一起使用时,Illumina表现出最高的覆盖率,因此具有最佳性能。但是,需要对此现象进行更详细的研究。有趣的是,对于Illumina较旧的平台Genome Analyzer II,已证明某些错误与反向重复有关[ 22 ],并且已知人类基因组包含大量双向启动子[ 34]。]用于相邻和相反方向的基因。已知这些双向启动子在其中点附近具有互补和对称的碱基序列内容,并可能以反向取向的转录因子结合位点的形式包含反向重复序列[ 35 ],可以很容易地用现有数据进行测试。
读取数据的纠错
现在,我们提供了高通量测序错误校正软件的方法学概述,该软件将读取的数据作为输入并输出校正后的读取。我们关注的工具是可以独立运行此过程的工具,而出版物或文档则提供了该方法的详细信息(表3和表4,补充表S2)。但是,我们还列出了纠错只是很多步骤中的一个步骤的软件,例如序列汇编程序和单元型推断工具ShoRAH和KEC,尤其是当它们贡献了重要的纠错思想或纠错工具可以独立运行时。最初,我们概述了计算方法,即使用了哪些数据结构,以及算法如何生成和操纵它们以进行错误检测。然后,我们讨论所使用的错误模型,包括它们所表征的特征,以及统计方法,包括所使用的模型及其参数。重要的是,我们重点介绍了由不同工具做出的假设,并指出哪些假设适用于哪种数据类型,以便于针对特定环境的基准选择工具。大多数高通量测序错误校正方法都基于三个主要假设:首先,在有足够覆盖范围的情况下,与正确碱基检出相比,错误(每个位置)被认为是罕见的。其次,覆盖范围在整个查询序列中被视为均匀。第三,替换和插入缺失错误都被认为在每个序列模板位置具有相似的概率。如果只知道某些数据类型的整体错误率,则这些假设可能足够合理。但是,考虑到还存在影响覆盖率和错误频率的系统错误(偏倚),因此,更复杂的方法可以更充分地检测并消除序列数据中的错误。我们首先描述基于这三个假设的方法,
在查询序列位置上堆积读取:通过读取比对,k- mer计数或读取后缀确定基频
为了利用高通量(高通量测序可达到的)覆盖范围进行纠错,必须根据序列读取所依据的基础序列的位置对它们进行分类。这可以使用多种技术来实现,具体取决于参考序列是否已可用,并导致碱基堆积或比对柱。然后,检查具有多个发散碱基调用的位置,以区分真正的多态性与测序错误-利用错误很少且随机的假设。在序列位置上确定碱基频率的第一步是通过比对读数或通过检查定义长度k(k-mers),其中后者可以使用某种形式的k -mer计数表或后缀数组来完成。从[ 36 ]改编为基于比对或基于k -mer的方法的分类。
读取比对:参考映射和多序列比对
只要有参考基因组可用,就可以通过找到每个读取到该参考序列上的最佳图谱来完成每个序列位置的碱基堆积(图3 A)。但是,通常没有(良好的)参考基因组可用或与参考的(大量)差异,这将导致比对中的参考偏差,从而导致位置堆积。在这种情况下,无参考组装或阅读分组策略可用于生成阅读的多序列比对(MSA),从而生成位置堆积。
从历史上看,此类无参考方法最初是为组装Sanger测序数据而开发的,这种方法产生的读数很少,读段较长,覆盖率较低。使用如此小的数据集,所有读取对的成对比对(例如[ 37 ])是可行的,并且从这些成对比对中,可以轻松构建和完善MSA(图3 B)[ 38]。]。有了来自下一代测序平台的更大数据集,所有可能的成对比对的初始步骤就变得难以计算。取而代之的是,较新的方法首先确定共享子序列的读对,然后仅对那些对进行比对,从而大大减少了执行的比对次数。为此,他们借鉴了以下我们讨论的其他三种堆积方法之一:通过创建一个索引表,其中包含指定长度k的所有读取子串k-所谓的k -mers(“ k -mer频率和频谱”部分) ) -和在其中记录读取它们发生(灰色读出的索引表在图3中; [ 39,40]),通过这样做,从A到B的引用映射(灰色虚线箭头图3 ; [ 41,42通过构造一个后缀数组])或(部分“后缀尝试和数组,Burrows的惠勒变换和全文索引分钟数”中的所有读数[ 43 ]或其派生词(图3中从F到B的灰色虚线箭头; [ 44 ])。
最终,MSA工具(补充注释S1)使用精制MSA的每个对准列中的基频来进行纠错决策;可以通过简单的多数表决,也可以通过“使用统计误差模型进行去噪”一节及其后的部分中讨论的更复杂的方法之一。
k聚体频率和频谱
并行地,开发了一种基于k- mer频率的方法来确定位置堆积中的基本频率,并且该方法在纠错领域中占主导地位。这个想法最初是在2001年的Sanger读物的EULER汇编程序中引入的[ 45 ],最初,它主要与EULER的汇编程序版本一起发展。
对于MSA方法中成对读取比对的种子子序列,记录所有可用读取集中存在的每个k- mer。但是,除了存储读取k -mer的信息外,还不计算并保存读取集中k -mer的出现总数。这为每个k- mer提供了一个“覆盖率”(发生率)值(例如,图3和4中的k- mer计数表)。使用的假设是错误是罕见的,随机的,一个阈值被选择,然后低频区分ķ被认为“错误”(或“弱”或“不可信的”)和高频聚体ķ-mers(“稳定”,“强”或“可信任”;图4中的绿色计数)-这近似于被测序样品的实际“ k -mer光谱”(图4 C)。原来ķ聚体频谱校正方法是在同一时间检查一个完整的读取,并认为这是错误的,如果它至少包含一个不受信任的ķ聚体,换句话说,如果ķ读的聚体光谱包含ķ -不在完整序列样本的可信范围内的用户。可以通过将所有不可信k变为必需的最小基本编辑次数来纠正这种读取-mers成为受信任的读物(另请参见“仅替换与替换加indel:汉明与Levenshtein距离”一节),或者,如果无法做到不超过所讨论读物的最大预定义更改,则将其丢弃。使用k- mer谱的许多不同的纠错方法(已经开发了补充注释S2)或仅k- mer频率。我们还将针对频率阈值和k- mer长度的选择进一步讨论这些内容(“信任k -mers的全局频率阈值”和“最佳k- mer长度”部分)。
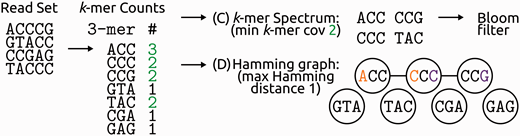
{kind=link}
导出ķ聚体谱或从一个汉明图ķ -mer计数。一些纠错工具直接使用从读取集计数的k- mer频率工作。其他人设置了最小k -mer覆盖率(在此示例中为2,绿色),以将k -mer视为正确(可信k -mers,绿色计数),然后得出所有可信k -mer的(C)k -mer谱。。在此简化示例中,此步骤将查询序列末尾的k -mers分类为不可信(可以将图3 A中的引用视为四次读取查询的序列)。通过使用布隆过滤器,可以降低k聚体谱。k- mer方法中使用的另一个概念是Hamming图,其中节点是读取集中的k- mers,如果Hamming距离(即它们之间的碱基取代数目,则连接节点);另请参阅“仅替换与替换”部分加indel:汉明与Levenshtein距离')低于给定阈值。在此简化示例中,k -mers太短,因此汉明图将三个正确的k -mers连接起来。在实际的设置中,ķ聚体长度必须小心(部分“最佳k链节长度”)和汉明图的最连通分量来选择应该只包含一个单一的正确ķ-mer加k -mers从相同序列中生成,但有错误。该图的彩色版本可从BIB在线获得:http://bib.oxfordjournals.org。
后缀try和数组,Burrows Wheeler变换和Minute空间中的全文本索引
确定每个序列位置基本频率的第三种流行方法是使用基于读取后缀的数据结构:后缀try,后缀数组及其派生词(图3 E,3 F,5和6)。后缀特里结构,在(混合)SHREC [使用46,47 ],是所有读操作,其中,每个边缘由数量加权的所有可能的后缀树读取支持它(图5)。虽然后缀特里需要大量存储空间(由测序遗传材料的长度,读取的长度和错误的数量决定),但它允许对整个数据集进行非常快速的字符串搜索。同样,权重枚举了在树中直到该级别为止具有相同前缀的读取后缀的数量,如果我们将检查到的树的级别视为一个k- mer的长度k,则trie直接提供所有可能的频率k- mer长度从1到最大读取长度。但是,并非所有的trie级别都可用于纠错,并且仅检查了trie的中间级别以检查每个节点处的边缘权重是否不平衡(以讨论k中的最佳点)。-mer长度,请参见“最佳k -mer长度”部分,补充说明S3)。

{kind=link}
后缀树是索引示例读取集中所有后缀的树。可以使用从根节点到读取索引之一的路径来拼写每个现有的后缀,该路径由箭头和相应节点处的读取编号指示。特里边缘的数字对应于通过它们的后缀的数量,即,边缘权重给出了从根到下一个节点的序列覆盖范围。例如,在四个示例读取中,2聚体“ CC”发生了六次。
后缀数组[ 48 ]首先由HiTEC [ 49 ]使用,是所有可能的读后缀的排序数组,通常与辅助索引结合:最长的公共前缀(LCP,图6 F)。对于后缀数组中的给定位置,LCP给出各个后缀与数组中其后缀共享的最长公共前缀的长度。使用这两个数据结构,它们所需的空间比后缀trie少得多,因此可以像后缀trie一样轻松地查询k mer频率,以查询不同长度的k mers(HiTEC中的“见证人”)。HiTEC利用此属性来迭代各种k选择用于优化校正的链节长度(“最佳k链节长度”部分和补充说明S3)。最近针对PSAEC [ 51 ]和Fiona [ 52 ]中的后缀数组实现了一种最先针对后缀树[ 50 ]引入的优化思想:在并行化的过程中,它们仅构造高达指定阶数h的部分后缀数组。这有效地意味着后缀仅根据它们的前h个基数进行排序,这仍然允许确定直到h的所有k- mer长度的k- mer频率。

{kind=link}
首先从运行的示例读取集中得出后缀数组,然后得出BWT和FM索引的步骤。还给出了一个使用BWT和FM索引进行字符串搜索的示例,紫色,红色和绿色分别跟踪相应的索引和核苷酸。对于后缀数组的构造,每个读操作都附加一个唯一的终止符号($ x),并且读操作按其终止符号的词典编排顺序连接到字符串R($ 1 <$ 2 <$ 3 <$ 4)。所有可能的后缀都明确地形成和排序,因为终止符号具有顺序,其他符号$ <A <C <G <T也是如此。后缀数组索引i处的后缀数组条目对应于字符串R中的位置,在我-th(按字典顺序)最低的后缀开始。然后,记录后缀数组条目和前一个条目的LCP,并且后缀数组加LCP已经形成了用于确定字符串出现频率的有效数据结构。BWT可以进一步压缩数据。在此,BWT索引i处的条目对应于R中第i个最低后缀的符号。与FM索引一起,它可以在整个读取集中进行线性时间字符串搜索(因此也可以确定字符串的覆盖范围)。FM索引给出了每个符号的出现次数,最高可达BWT的任何索引,并且对于每个符号,对所有按字典顺序排列的较低符号的出现次数进行计数(例如,对于符号“ G”,出现的次数为4 + 4 + 10 = 18,蓝色)。该图的彩色版本可从BIB在线获得:http://bib.oxfordjournals.org。
在SGA汇编器中[ 44 ],采用了一种不同的优化数据结构,即Minute空间中的全文本索引(FM索引;由Ferragina和Manzini发明;图6 H; [ 53 ])用于纠错-一种数据结构。最初是为从头装配中的弦图构造引入的。FM索引结合两个辅助数组使用Burrows Wheeler变换(BWT,后缀数组的无损压缩;图6 G; [ 54 ]):BWT中每个符号的累积出现次数按以下顺序计数: BWT索引(图6中的Occurrences表)H),并记录在字典上低于完整BWT中某个碱基的所有碱基的总数(图6 H中的五个计数,每个符号一个计数,按字典顺序累加)。在组装费米[ 55 ]中,FM索引得到了进一步优化,以将DNA的反向互补链都包含在一个索引中,即FMD索引,它允许在模式搜索中进行双向匹配扩展。
错误检测和纠正:从错误中分辨出真正的多态性
仅替换与替换加上插入/替换:汉明与莱文施泰因距离
代替允许测量单个核苷酸插入,缺失和取代的Levenshtein编辑距离[ 56 ],大多数错误校正工具使用Hamming距离[ 57 ],该距离仅考虑取代。这通常由两个主要论点证明:第一,替换错误的数量比主要Illumina数据中的indel错误高一个数量级;第二,仅使用汉明距离的方法的计算复杂度较低,这是对于高吞吐量数据中的错误纠正程序和从头组装等计算密集型任务尤其重要。结果,大多数工具只能纠正替换错误(表3),甚至有人将汉明距离作为中心概念:k -mer频率工具Reptile [ 58 ],Hammer [ 59 ]和BayesHammer [ 60 ]创建一个汉明图,其中k -mers作为节点通过边连接,如果它们之间的汉明距离低于某个非常低的阈值(图4 D)。通过在所有k- mer列表的不同副本中进行邻近搜索,可以很容易地找到这样的连接,每个副本均被系统地分类,而忽略了一定数量的位置,其中,每个副本的分类中忽略的位置数量决定了允许的汉明距离。MSA工具Coral [ 61]和ECHO [ 62 ]是邻接表的使用。最后,FreClu [ 21 ]和Aita等人的未命名工具。[ 63 ]聚类按汉明距离进行读取,然后对所得邻域(即聚类)使用不同的纠错策略。
另一方面,只有很少的工具(表3)明确实现了indel校正:在MSA工具中,这可以通过使用允许间隔的成对对齐算法创建或优化MSA来实现(例如[ 37 ])。在大多数MSA错误校正工具中就是这种情况,只有一些较新的工具以indel校正能力换取计算速度:SGA [ 44 ]使用优化的数据结构(后缀数组的BWT表示,图6),并从中创建MSA。它使用本身不支持indel检测的算法。ECHO [ 62 ]尚未实现插入缺失校正以节省计算资源。
Fiona [ 52 ]中实施的一种最新方法是,通过从错误的和所有相应的正确读数之间的后缀数组种子中找到最佳的比对扩展,来优化三种不同单核苷酸错误类型的校正可能性。这在错误位置处实现了Levenshtein距离,甚至允许对齐扩展中进一步插入或删除,然后在优化中受到不利影响。
通常,由于Ion Torrent,PacBio和Oxford Nanopore出现的新兴平台会产生大量的indel错误(表1和图2),因此决定是否实施indel错误的纠错变得越来越重要。尽管最近的k- mer工具(例如Blue [ 64 ])明确地考虑了indels,尽管计算复杂度有所提高,但仍可以进一步举例说明。
信任k-mers的全局频率阈值
许多k- mer频率工具通常通过最小化它们之间的汉明距离来创建k- mer频谱,以将不信任的k- mer校正为最接近的可信任的k- mer(即,仅替换,'仅替换与替换加插入/插入缺失:汉明与Levenshtein距离')。为此,他们使用这样的假设:错误很少发生,不同的错误发生的可能性相同,并且覆盖范围是一致的,并依靠频率阈值来决定信任哪个k聚体(例如,图4中的两个)。而这个阈值最初是手动从经验-例如选择在欧拉[ 45,65 ]和SOAPdenovo [ 66,67]-很快就建立了从客观数据中得出阈值的客观标准(表3,补充表S2)。
在EULER-SR中,阈值是手动选择的,但是提供了选择的明确理由[ 68 ]:给定读取次数(N),平均长度(L),近似基因组大小(G)和k -mer长度(在原始出版物中称为l),平均k -mer覆盖率可以计算为:a = N *(L − l)/ G。然后,假定正确的k -mers覆盖率遵循泊松分布。围绕平均一个,并且选择覆盖范围阈值,以使理论上只有极少数正确的k- mers(例如,在整个数据集中少于100 k- mers)会降至该阈值以下。沿着相似的路线,ALLPATHS [ 69 ]假定经验分布的基础是两个不同的分布:一个用于错误的k- mers(频率非常低,图7),另一个用于正确的k- mers。一致地发现,这两个分布的峰均被经验总体k- mer分布的第一个局部最小值隔开,因此,将该第一个局部最小值作为阈值(图7)。)。随后开发的大多数工具都采用了类似的策略,并对其进行了一些改进(有关确定各个工具的全局k频率阈值的细微差别,请参见补充说明S4)。在这方面最值得注意的是Quake [ 70 ]:此处, k- mer频率由质量值加权(产生“ q- mers”),以更清楚地分离经验分布最大值。另外,第三分布适应来自在查询序列中的重复的高多重性k聚体的重尾(图7)。Quake将这些重复的k -mer投影到分布中以获得正确的q -mer,然后使用其完整混合模型的最大似然拟合来确定q -mer频率阈值(补充说明S4)。

{kind=link}
具有模型拟合的k- mer覆盖直方图。在这张来自Quake论文[ 70 ]的图中的直方图给出了一个经验性的k- mer覆盖分布的很好的例子。密度告诉我们数据集中所有现有k- mers中有哪个比例具有特定的覆盖率。实线表示Quake模型拟合。分布的第一个峰值是由极低的覆盖误差k- mers形成的,通常通过泊松或伽马分布进行建模。第二个峰值是由大多数正确的k- mers产生的,通常通过泊松或高斯分布来建模。在这两个峰之间,一个清晰的局部最小值可以提供一个k-mer信托承保范围截止。较高k -mers的重尾是来自序列重复的k -mers的结果。Quake从Zeta分布中提取了k -mers的序列拷贝数,然后将这些k -mers投影到正确的k -mers的覆盖范围内。根据知识共享署名许可CC-BY 2.0(http://creativecommons.org/licenses/by/2.0/),根据[ 70 ]中的字体更改和标签添加进行了改编。
依赖可分离k- mer分布的方法的一个主要警告是,它们仅在覆盖范围均匀地分布在查询序列上时才适用。因此,此处提到的纠错工具(以及任何具有全局k- mer信任阈值的工具;表3)不适用于具有固有变异覆盖率的数据集,例如在宏基因组学和转录组学中,或者在覆盖率存在严重偏见的情况下(例如,由于(例如在单细胞测序中)所使用平台的GC偏倚或由于样品偏向全基因组扩增的倾向。要更正此类数据集,可以考虑“消除覆盖范围假设的一致性”部分中介绍的软件。
最佳k聚体长度
软件使用ķ聚体的频率有选择一个特定ķ聚体的长度(例如,三个在图3,4和8)。在描述Quake [ 70 ]的论文中,首先详细讨论了这种k长度选择所固有的权衡(对于de Bruijn图(图8)组装者来说同样重要的问题):' k的较小值提供了更大的判别力用于识别读取中错误的位置,并使算法运行得更快。但是,k不能太小,以至于很有可能一个k单核苷酸取代后,基因组中的k- mer与基因组中的另一个k- mer相似,因为这些情况会混淆错误检测。”,即每个k- mer必须足够长才能在查询的序列中是唯一的(假设每个k -mer都可能在整个序列的任何位置发生),但是如果某个工具只能检测到每个k -mer错误,这是大多数工具的默认设置,因为它们在k -mer之间使用汉明距离为1 —较长的k- mer表示较低的错误解决方案。为了确定长度,通常要求用户提供一个k-mer长度(有时通过SGA的“ sga stats”功能提供某些指导; [ 44 ])或软件提供的默认值在实践中很有效(表3)。相比之下,Quake的作者更系统地分析了上述权衡取舍,并建议以下设置k -mer长度:'[T]从(4)的空间中随机选择k -mer k的概率^ ķ)/ 2 [...]可能ķ发生在核苷酸的随机序列的测序基因组的大小聚体G ^,是~0.01' 。基于这一要求,他们给出的近似值ķ ≈log_4(200 *G)。对于大约5 Mb的大肠杆菌基因组,建议的k- mer长度为15,对于大约3 Gb的人类基因组,建议的k- mer长度为19。但是,更长的k- mers可用于更好地解决纠错中的重复,并且现在变得可管理:平均而言,更长,更准确的读取平均每个单次读取会产生更多正确的k- mers;更深的测序给出足够高的ķ对较长校正决定聚体覆盖率ķ聚体(否则一个问题,尤其是对于组件邻接[ 71 ]); 较新的实现方式和硬件可以处理生成的k- mer光谱。
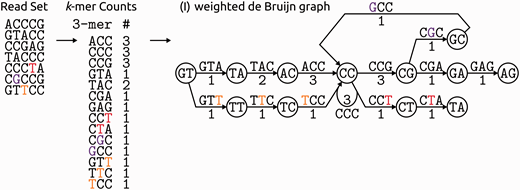
{kind=link}
示例阅读集中的加权de Bruijn图的示例。与早期图中的示例读取集相比,该读取集得到了增强,以包括显示出差异的读取:尽管T(红色和橙色)可能是替换错误,而G(紫色)可能是插入错误,但所有这三个也可能是读取覆盖相同序列基因座的替代等位基因或在其他序列基因座处的重复稍有不同的重复。新的读取创建了通常在图修剪(并因此进行校正)步骤中删除的图结构:(i)橙色T产生了一个凸起,即该图中的一个循环,该循环不能被单个路径作为循环的边缘进行遍历朝相反的方向走(例如,边缘ACC和TCC都在节点CC处结束);(ii)红色的T创建了一个尖端,即该图的短死角;(iii)紫色的G产生旋转感,图中的一个循环,其中可能的路径围绕整个循环(即,所有边沿相同方向)。重复图将消除小(并且可能是错误的)漩涡,隆起和尖端,而重复图会崩溃,但是必须注意不要消除真正的变异。该图的彩色版本可从BIB在线获得:http://bib.oxfordjournals.org。
有明确动机的k- mer长度选择的其他示例来自后缀和数组的领域(图5和6),其中数据结构本身允许检查几个不同的k值。(混合)SHREC [ 46,47 ]只在树的中间水平检查节点的权重,对应于中间ķ聚体的长度,其中,树的高度由所读取的长度来确定。HiTEC [ 49 ]确定最小化假阴性校正概率的k值和最小化假阳性校正概率的k值。然后使用多个k-mer长度围绕这两个最优值。Fiona [ 52 ]也采用了这种方法,并对其进行了进一步调整,以适应变化的读取长度。总之,这些方法自动确定ķ聚体长度他们使用和较不易于ķ因为它们使用多个聚体长度效应ķ聚体的长度的修正运行内(详情补充说明S3)。
最后,最近一个最初的目的是确定用于与de Bruijn图装配的k- mer长度的工具称为KmerGenie [ 72 ],它有可能也自动确定基于k -mer的误差校正的最佳k- mer长度。它使用Quake的k -mer分布模型的扩展,使其分别适合于不同k值的经验k -mer计数分布。该模型拟合估计每个k的不同正确k -mers的数量(即,该长度的k -mers将在正确的参考中)和k正确的k -mers数量最多的对象被认为是最佳的。为了使ķ为聚体计数经验ķ聚体分布计算上易处理为的多个值ķ(关于两个运行时和存储器消耗),KmerGenie子样品的ķ通过因子聚体ε(作者使用ε = 1000)。
消除覆盖范围假设的一致性
如以上所讨论的,如果要信任k- mer计数,则k- mer方法主要依赖于全局阈值,并且该阈值做出非常强的假设,即,在整个查询序列中覆盖范围是统一的。因此,无论阈值确定多么复杂,该方法都会将来自确实存在于查询中的低覆盖范围区域的k个聚类错误地分类为不可信,并且同样会对具有固有变体覆盖范围的数据集失败。甚至不建议根据经验k- mer频率分布自动确定全局阈值,以防止Quake [ 70],并且通常会失败:在覆盖范围不均匀的数据中找不到正确的和错误的k- mers(图7)可辨别的直方图峰。
因此,一些更新的基于k -mer的工具避免使用这样的全局阈值,而使用以下策略之一:
-
Reptile [ 58 ],Hammer [ 59 ],BayesHammer [ 60 ]和Sleep等人的工具。[ 73 ]通过创建k -mers的汉明图,按汉明距离对读段进行隐式分组(关于汉明距离和汉明图,另请参阅“仅替换与替换加插入缺失:汉明与Levenshtein距离”一节和图4 D) 。在汉明图中,他们检查了非常相似的k- mer的连接组件,称它们为k- mer邻域。基于Hamming社区内k个单体的相对频率,Hammer选择一个共识k-mer用于每个连接的组件,因此对每个汉明邻域做出一个本地决策,并输出校正后的k -mers。BayesHammer通过k-均值聚类进一步细分汉明邻域,其中子簇的概率近似于所含读段的测序质量得分。此子集群区分了非常相似的k- mers,它们最初共享一个邻域,但起源于不同的序列位置(例如重复序列),但不应从错误的k- mers创建新的子集群。然后创建一个k-高品质簇的质谱图,基于高品质读数对其进行扩展并进行基于读数的校正。爬行动物在其纠正方法中也将阅读作为一个单元。它检查每个读取的多个k聚体,以它们称为“平铺”的顺序指定顺序,在各个汉明邻域内搜索频率较高的k聚体,最后将读取结果更正为替代平铺(一组替代k -mers)(如果它具有比原始读取的k- mers的相应集合更高的覆盖率)。因此,此校正决策比Hammer集成了更多的上下文信息,并且至少在本地确实依赖更统一的覆盖范围。
-
蓝色[ 64 ]使用k -mer谱(最初使用非常低的全局排除阈值),但是创建其k -mer信任阈值以分别校正每个读取。以这种方式,它使它适应于局部覆盖,并允许读取之间的k- mer覆盖变化。
-
修平刀[ 74 ]根本不使用覆盖阈值用于其k聚体谱。取而代之的是,它最初仅包含k -mers,其碱基的质量值至少在一次读取中位于数据集的前8%之内,然后通过“增强”校正后的碱基的质量值到最大质量值来迭代扩展光谱以校正k -mer而著称。这样,根本就不会做出覆盖假设。
-
像BayesHammer,Reptile和Blue一样,EDAR [ 75 ]也以其校正方法一次专注于一次读取。它对每个读取的区域进行分类,从而规避了全局阈值。使用可变带宽均值移位法,它识别与一致沿读取连续区段ķ聚体覆盖率和识别断点其中覆盖快速改变到一个不同的一致的水平(用的GC-偏差校正的独特特征ķ - mer频率)。然后,它对读取区域进行分类,而不是k-mers或full reads-不受信任(覆盖率非常低)或受信任,并将受信任区域进一步细分为唯一和重复区域(覆盖率非常高)。这是通过固有的本地聚类决策来完成的([ 75 ]中的图1)。
-
一次不查看完整的k- mers,而仅查看堆积的一个基本列,也就完全避免了全局阈值:可以在不假设全局覆盖均匀性的前提下,根据各个位置的相对基本频率进行决策。QuorUM [ 76 ]就是这样做的:它使用依赖于每个位置的基本频率的改进的投票方案。ALLPATHS-LG [ 77 ]和fermi [ 55 ]通过原始阅读的相关质量得分进一步加权这些频率,使Quake的“ q- mer”思想适应各个堆积位置(与“信任k的全局频率阈值”部分比较)-mers')。
对于MSA方法来说,需要特别关注单个位置而不是k - mer工具中的k- mers变得更加自然:可以轻松避免全局阈值,因为可以直接根据每个基本列的相对频率做出决定对齐方式。然而,为简单起见,SGA的[ 44 ] MSA模块仍然使用全局阈值,但是所有其他MSA工具都进行某种基于列的多数表决或统计测试,从而有效地采取了真正的本地决策。
最后,两种基于隐马尔可夫模型(HMM)的纠错方法SEECER [ 78 ]和PREMIER [ 79 ]也固有地采用局部决策,其发射概率分别来自MSA对准位置或k- mers。
总之,这里描述的软件对于测序范围不均匀的数据集(例如转录组学,宏基因组学和单细胞基因组学)特别有趣。但是,不管有多么重要,要避免假设统一的覆盖范围,采取局部决策(通常一次在一个查询位置)通常会忽略上下文信息。在本节中,我们已经在BayesHammer,Reptile和EDAR的整个阅读范围内瞥见了此类上下文信息的使用。但是,在“重复和单倍型模型”部分中,对工具如何在每个检查的位置周围使用更长的上下文范围进行更正决策进行了更为详尽的说明。
使用统计误差模型进行降噪
大多数工具根本没有错误模型,而其他工具则可以解决一些我们已经审查过的错误偏差,以便更精确地区分低频下的测序错误和真正的序列变异。最简单的误差模型使用全局(即均一)错误时经验已知为各个测序平台或采取分配PHRED质量分数作为用于局部误差概率(例如,[取代概率60,63,74]),因为事实证明它们通常与大多数平台的测序错误相关。但是,正如我们所回顾的那样,已知错误频率会因平台的具体情况(不同的碱基混淆概率和整个读取过程中的信号衰减),局部序列内容(GC内容或序列基序,例如均聚物延伸和反向重复)而有偏差。测序方案之前的步骤(例如,预扩增)。
因此,几种工具使用的方法(补充说明S5)将采用根据经验确定的基本混淆矩阵,该矩阵单独给出每种可能的碱基取代的概率,而不是假设统一的取代概率(即具有4×4取代概率的矩阵)。在这里,可以使用各个平台的通用矩阵,但是大多数工具会从数据中学习矩阵并合并其他功能(补充说明S5)。454焦磷酸测序的类似概念(其中均聚物长度错误是最突出的错误类型)是使用每均聚物长度的光强度分布,该分布是根据已知序列的测序经验确定的,例如Balzer等人。[ 18 ]。
几种工具还使用其他特定的方法来估计错误:AutoEdit [ 80 ]将来自读数比对的信息与其原始毛细管测序流程图的峰分辨率的三个度量结合起来,以确定碱基检出是否正确的可能性。RECOUNT [ 81 ]通过获取该位置对齐列的质量值平均值,计算每次读取中每个位置的错误概率。pacbio_qc [ 28 ]旨在通过PacBio RS平台针对CCS,将读取次数(CCS的一项特殊功能)和平均质量值汇总到错误概率中。
最后,三个工具使用了更精确的错误模型,这些模型值得更详细的介绍:
-
SysCall [ 82 ]的作者首先分析了错误发生,并在使用Illumina平台时确定了一种新型的系统错误。这些错误表明:(a)与周围碱基相比,覆盖率下降;(b)错误位点处的碱基特异性混淆矩阵非常明确,并且两个碱基在上游直接偏向;(c)强烈的链偏向。基于这些特征,他们使用从已知序列中读取的数据作为训练数据,训练了逻辑回归分类器,以区分真正的杂合子与潜在杂合子位点处的此类错误。
-
PREMIER [ 79 ]拟合数据中所有读取的k- mer转移概率的HMM 。对于给定的发射k -mer,对于读取中的某个位置,转换到下一个位置的概率是从四个值得出的:(a)读取中的位置,(b)替换错误的概率给定该位置处的质量值,则在该位置处,(c)通用基本混淆矩阵,以及(d)数据中可能的排放量k- mer的计数。但是,作者没有提供有关运行时的任何基准测试,这表明该方法可能无法扩展到更大的测序数据集。
-
Fiona [ 52 ]使用排序过程的分层统计模型来确定位置是否错误。使用均匀的覆盖和均匀差错概率的假设,它确定给定的观察到的一个错误的数比值比ķ聚体的特定的可能错误的覆盖ķ聚体,并且在任何一个错误的通用日志比值比ķ -梅尔 如果特定k -mer中错误的对数比值比一般k -mer中错误的对数比值高,则该k -mer被认为是错误的。补充说明S6)。如作者所述,如果找到更合适的阈值或需要不同的纠错灵敏度,则可以轻松更改此阈值。但是,不仅可以轻松调整模型的这一方面,这使得该模型成为将来开发更灵活的纠错工具的有吸引力的起点。
总而言之,许多错误校正工具通常不会出于统计目的对错误进行建模(通常是为了简化起见)。同时,这里介绍的少数几种确实包含某种错误模型的工具在其方法上显示出相当大的多样性,大多数模型的核心都是某种形式的标准或学习型基础混淆矩阵。尤其是在像Illumina这样的平台上,错误已经得到很好的表征,读取错误纠正可以从这种更具体的方法中受益。
重复和单倍型模型
测序错误校正中的中心问题是将错误与真正的变异区分开来,无论这种变异是由于基因组内的重复,还是由于群体或单个细胞内的不同等位基因或单倍型所致。大多数工具都是基于这样的假设,即错误很少且是随机的,并且覆盖范围是一致的,而其他工具则在其校正程序中忽略了来自重复区域的读数或k- mers,例如Fiona [ 52]。]。我们描述了四种对真实变异的预期足迹进行建模以使决策更加清晰的方法:(i)ECHO对二倍体基因组进行建模;(ii)EULER-USR使用重复图提供重复模型;(iii)MisEd,SGA,Acacia和SEECER都确定读段和共有序列之间的错配是否在读段内链接在一起,以消除不精确的重复序列;(iv)ShoRAH使用Gibbs采样器将读数分配给任意数量的单倍型。
-
ECHO [ 62 ]给出了特定阅读位置上每个可能基因型的后验估计,假设该位点是杂合的(相对于纯合的)先验概率,并且给出了包含阅读位置的MSA的碱基频率。对于单倍体基因组,该概率可以简单地设置为0。因此,此模型有效地允许在每个检查位点使用一个或两个单倍型。
-
EULER-USR [ 83 ]使用其程序集数据结构,即重复图,以使其能够进行重复纠错。首先,使用简单的k- mer光谱方法对读取前缀进行了纠错,因为它们被认为是读取的最准确部分(在大多数平台中,错误率随读取长度的增加而增加,请参见“测序平台及其错误”一节)。然后,从更正后的读取前缀构造一个A-Bruijn图:这是de Bruijn图的广义版本(图8),其中连接相同顶点的所有边汇总为一个边,其权重反映了原始数目覆盖它的边缘[ 84 ]。消除了不一致的图结构(即凸起,尖端和旋转,图8),这有效地消除了图中的测序错误,简化图中唯一剩下的缠结应该是真正的重复(或单倍型),从而得到重复图[ 84 ]。然后,可以通过将整个读取映射到此读取前缀重复图并将其校正为共有值来校正不太准确的读取末端。最近的PacBio混合错误校正器LoRDEC [ 85 ]也采用了类似的方法:在这里,长错误倾向的PacBio读取通过将de Bruijn图穿入线程中进行校正,该图由来自不同平台的更准确的短读取构成(请参见本节“平台特定的错误纠正”)。这种基于图的纠错方法及其图抽象-由de novo设计 考虑组装而不是人口重新排序-模型在基因组中隐式重复(或杂合),但最有可能在去除低覆盖率多态性方面出错。
-
MisEd [ 86 ]是第一个利用以下事实的工具:给定足够长的读取时间,源自(不准确)重复的真正差异将始终连贯地存在于多个读取中,而错误(假定很少发生且随机发生)应该被隔离单次读取或至少减少一次读取(图9)。为此,他们使用了以前开发的定义核苷酸位置(DNP)的概念; [ 87]):偏离(MSA)共识的列被视为候选,并且对候选对进行读取内显着的同时发生测试,即,鉴于涵盖两个区域的读取次数,它们近似于两个位点之间的预期同时发生量站点,并根据Poisson分布对站点上的相应质量值进行建模。据此,他们计算观察到至少观察到的,共有两个偏差且与共有序列有偏差的读数的概率,如果该概率超过预定义的阈值,则将两个位点视为DNP。MisEd对相对于MSA共识具有更高预期失配量的区域进行DNP分析,并通过共识调用MSA的每一列来进行后续的纠错,从而跳过了已识别的DNP作为真正的重复序列。因此,MisEd使用一个明确的重复模型,该模型可以保护所有可以识别的重复(或单倍型)变异免遭假阳性错误校正。SGA组装器的MSA模块[[44 ]使用类似但更简单的校正前检查:它将MSA中冲突的列标识为至少两个不同基数超过给定最小覆盖范围阈值的列。然后,如果它们在所覆盖的所有冲突列的一致性始终不一致,则在进行错误纠正之前会从原始比对中排除读取,从而有效地合并了比MisEd更简单的重复模型。金合欢[ 88]是为454个焦磷酸测序数据开发的工具(“平台特定错误校正”部分),也使用SGA中的错配之间的链接。但是,它会对游程长度编码的读段进行聚类和对齐(即折叠其所有均聚物拉伸),然后识别读段之间均聚物长度上有显着差异的对齐列(“平台特定错误校正”部分)。在整个比对中共有多个这样的均聚物长度差异显着的读段将被分成一个新簇,该簇被反馈回原始过程。SEECER [ 78]是主要针对RNAseq数据的工具,其工作原理类似:它通过在初始重叠群中分离不同的不精确重复序列(或单倍型)来完善MSA重叠群。通过共享各自不匹配的一组读数来识别和聚集与MSA共有序列不匹配的列(谱聚类和k均值的谱弛豫)。通过这种方法,可以将读数的一致和均质的子集分离出来,以进行进一步的分析,因此,聚类暗示了重复和单倍型的模型(更多SEECER详细信息请参见补充说明S7)。
-
ShoRAH [ 41,42]针对包含相同物种许多单体型的异质样品,如准物种的病毒群落。它在映射派生的MSA中检查定义长度的重叠窗口(选择以使每个位置都被三个不同的窗口覆盖),然后重建其(局部)单倍型。为此,使用Gibbs采样器将该窗口中的读数聚类为单倍型,以从Dirichlet过程混合物(DPM)的后验分布中反复提取。DPM假定将读取分配给现有单倍型或新实例化的单倍型的先验概率。围绕此先验,它构建了一个单倍型模型,有效地生成了以下多元变量后验分布:(a)单倍型的读分配,(b)单倍型的序列,(c)正确碱基调用的每位概率(即一个减去在任何随机位置发生测序错误的概率),以及(d)单体型的每位概率与该位置的参考相同(也就是说,减去参考和单倍型在任何位置发生突变的可能性)。该迭代采样最终导致代表单倍型的簇的稳定种群,然后通过对与每个MSA位置重叠的三个不同窗口集的单倍型重构进行多数表决来进行校正。总之,ShoRAH中的聚类显式地模拟了任意数量的单倍型。一个减去在任何随机位置发生测序错误的概率),以及(d)一个单元型在该位置与参照相同的每个位置的概率(即,减去在任何位置的参照和单元型之间发生突变的概率)位置)。该迭代采样最终导致代表单倍型的簇的稳定种群,然后通过对与每个MSA位置重叠的三个不同窗口集的单倍型重构进行多数表决来进行校正。总之,ShoRAH中的聚类显式地模拟了任意数量的单倍型。一个减去在任何随机位置发生测序错误的概率),以及(d)一个单元型在该位置与参照相同的每个位置的概率(即,减去在任何位置的参照和单元型之间发生突变的概率)位置)。该迭代采样最终导致代表单倍型的簇的稳定种群,然后通过对与每个MSA位置重叠的三个不同窗口集的单倍型重构进行多数表决来进行校正。总之,ShoRAH中的聚类显式地模拟了任意数量的单倍型。该迭代采样最终导致代表单倍型的簇的稳定种群,然后通过对与每个MSA位置重叠的三个不同窗口集的单倍型重构进行多数表决来进行校正。总之,ShoRAH中的聚类显式地对任意数量的单倍型进行建模。该迭代采样最终导致代表单倍型的簇的稳定种群,然后通过对与每个MSA位置重叠的三个不同窗口集的单倍型重构进行多数表决来进行校正。总之,ShoRAH中的聚类显式地模拟了任意数量的单倍型。

{kind=link}
读集的MSA示例,与孤立的或低频的错配和插入缺失(红色的核苷酸和破折号)相比,显示出一致的错配(绿色核苷酸)。错配的一致性可以通过它们的连接(在该示例中为四个)被测试内读取和被称为在第一工具DNPS所使用这样的信息,MisEd [ 86,87 ]。但是,只有当多个链接的变体位点在平均读(对)覆盖的范围内时,才能从测序错误中分辨出真正的多态性。该图的彩色版本可从BIB在线获得:http://bib.oxfordjournals.org。
总之,在纠错工具中存在四种在相似序列中真实变异建模的方法:(i)ECHO中的二倍体基因组模型。但是,这既不适用于种群(的混合),也不适用于基因组内出现多个不精确的重复序列。(ii)EULER-USR(和LoRDEC)中的重复图模型。这可以容纳任意数量的不精确重复序列和/或单倍型,但旨在从头开始组装,因此可能会低估多样性,尤其是面对不同的单倍型或重复拷贝的覆盖不均匀时。(iii)MisEd,SGA,Acacia和SEECER使用一种隐含模拟不精确重复的方法来检查读物中的错配(与共识)的联系。当几个分别的单态多态性位于读段(或配对末端测序中的读对)所覆盖的距离内(好)时,这也适用于单倍型,但是将无法保护孤立的多态性(例如,类似的长重复序列)或来自人群中密切相关的品系)。(iv)ShoRAH实现了可以容纳任意数量单倍型的最灵活的单倍型模型。
平台特定的错误纠正
454焦磷酸测序
一些方法考虑了Levenshtein距离,因此包括对454个焦磷酸测序数据中普遍存在的indel误差进行校正。但是,甚至针对此类数据设计了一些工具,主要是针对扩增子测序的较小数据集。该领域的第一个工具PyroNoise [ 89 ]介绍了流程图聚类的思想。流程图记录了来自四个不同核苷酸在测序板上连续冲刷的光强度,并且可以通过将光强度四舍五入来确定各个核苷酸的连续积分总量(即均聚物长度)。但是,此过程容易出错,Quince等人。[ 89因此,返回到流程图以进行错误校正。他们将光强度的混合模型(单次冲洗)用于不同的均聚物长度,并从已知序列的测序中获悉概率分布。在此基础上,他们应用了期望最大化算法,以最大化从一组假定的真实序列(每个假定的真实序列一个簇)生成观察到的流程图(及其频率)的可能性。最终,将簇中最丰富的读段作为所有包含读段的代表。降噪器[ 90]通过使用启发式方法来构造初始簇来提高此方法的速度:它首先过滤掉作为其他reads前缀的reads,然后将按其丰富程度排序的已识别read前缀簇用作簇的起点。相比之下,AmpliconNoise [ 91 ]通过使用已知的碱基混淆矩阵,也考虑了扩增子生产的PCR步骤中引入的错误,从而使方法更加准确(补充说明S5)。
相反,还有其他三个工具在基准调用之后查看读取:Acacia [ 88 ],HECTOR [ 92 ]和KEC [ 93 ]。前两个都对均聚物使用行程编码(RLE)。折叠均聚物的想法变成了单个核苷酸,以允许在不考虑均聚物长度误差的情况下进行焦磷酸测序数据分析,最初是在Celera组装机的一种版本中提出的[ 94 ]。金合欢[ 88]散列RLE读取前缀(此时不考虑单个均聚物的运行长度),从而创建初始读取簇(让人联想到DeNoiser的预聚类)。然后通过合并具有相似的6-mer光谱的共有簇并通过从均聚物位点的游程长度与其余簇明显不同的读取集创建新的簇来精炼这些簇。最终,所有读物都将按照其簇的共识进行校正。HECTOR [ 92 ]也使用RLE,但不是将聚类方法应用于RLE序列,而是应用了k -mer谱方法(“ k -hopo谱”),包括自动确定k-hopo覆盖信任阈值从其经验分布(补充说明S4)。KEC [ 93 ]也使用k- mer频谱方法,但是没有以前的RLE编码。取而代之的是,它采用EDAR策略(比较“删除覆盖率假设的一致性”部分),该方法通过使用可变带宽均值漂移方法将读取的k聚体按其频率聚类来识别错误的读取区域。但是,KEC不会像EDAR那样仅仅删除已识别的错误区域,而是会考虑均聚物长度误差,对它们进行校正。
太平洋生物
从PacBio平台进行的长时间阅读中,非常高的总体错误率是主要挑战。当前,解决此问题的两种主要策略是要么使用来自另一个平台的错误较少的短读取,并具有足够的覆盖范围以纠正长的PacBio读取(称为混合方法),要么利用错误似乎在系统中没有偏见的事实。足够的覆盖范围,因此可以更正此平台。
第一种方法最初是在PBcR [ 95 ]中作为独立工具以及在组装程序AHA [ 96 ]中的流水线阶段实现的,后者是与机器供应商直接合作开发的。在这两种方法中,来自另一个平台的更准确的短读都会映射到长读上。然后,AHA只需将长读纠正为生成的短读映射的共识,而PBcR进一步优化比对,创建一个短读MSA,然后将其用于共识调用。LSC工具[ 97]通过使用RLE(即作者称其为均聚物压缩)的想法,改进了该方法的比对方法,该想法已在不久前用于454焦磷酸测序误差校正中(与上一节中的Acacia和HECTOR比较)。将RLE短读映射到RLE长读可忽略均聚物长度错误,从而提高了映射灵敏度。
相反,最近的一种工具proovread [ 98 ]改进了普通映射方法:首先,它通过使用适合于错误配置文件的对齐罚分值(对插入,删除,取代和缺口延伸的单独惩罚)使其更加敏感。其次,它通过并行化过程使其可扩展。它着眼于短读对个别长读的映射,并使用迭代的映射和校正过程,该过程逐渐包括更多的读,并允许在每一轮中进行更多不匹配的映射。第三,它识别并拆分嵌合的长读。
最近通过bioRxiv发布的ECTools [ 99 ]使用Celera Assembler将更准确的短读片段预组装为unitigs。然后,将长读与这些单位对齐,通过解决最长的子序列问题来优化此对齐方式,并朝单位进行校正。
LoRDEC [ 85 ]是另一种最近的混合方法,它也进行了预组装并借鉴了现有策略:通过广义的加权de Bruijn图进行线程读取的想法,最初是作为k- mer方法引入的,仅在短读时使用。 EULER-USR [ 83 ]。在EULER-USR仅使用更精确的短读词前缀构建图形的情况下(“重复和单倍型模型”一节),LoRDEC仅使用来自不同平台的短读词,因为它们比PacBio读书更准确。然后,EULER-USR将完整的短读数穿入图形以纠正它们(包括不准确的后缀)时,LoRDEC将长的PacBio读入线程。
对于非混合型PacBio纠错方法,PBcR被改编为使用来自PacBio RS测序运行的较高丰度的较短读数,以提供校正所有读数的覆盖范围,包括来自同一运行的较低丰度的较长读数[ 26 ]。这种方法也在当时称为HGAP的供应商组装管道中实现[ 100 ]。
牛津纳米孔
对于这项技术,到目前为止,仅通过bioRxiv出版物描述了一种工具:Nanocorr [ 101 ]。由于错误率与PacBio开发的早期阶段相似,因此它采用了类似的混合错误校正方法。在这里,BLAST用于将MiSeq Illumina的短读段与牛津纳米孔的长读段对齐。然后,类似于ECTools策略(参见上文; [ 99 ])对这种比对进行优化,最后,将短读比对的共识称为校正后的长读[ 101 ]。
结论
纠错工具的选择在很大程度上取决于您要执行的分析类型以及生成数据的测序平台,因为对于手头的数据集而言,特定方法的假设可能并不成立。一般而言,错误校正也已被证明对单核苷酸变异(SNV)调用[ 70 ]和单倍型重构[ 102 ]有益,但是该工具通常在基因组组装之前使用,以减少比对的复杂性和中间数据结构的大小。大多数最终用户仅依赖于他们选择的汇编器的内置纠错方法,因为几乎没有系统和独立的纠错工具基准可以证明选择的合理性(值得注意的例外是[ 36(103))。因此,我们认为许多不同的数据类型和应用程序都需要特定于上下文的基准,并提出以下六个方面来选择工具以对特定设置进行基准测试(表2和表3以及补充表S2):首先,最重要的是,该工具需要免费提供,此处提供的60种工具中有52种(表4和补充表S2)。其次,该工具是只需要考虑替代错误,还是应该考虑插入缺失,取决于生成数据的测序平台。对于Illumina和Complete Genomics数据,插入缺失错误的重要性不如454,Ion Torrent和PacBio平台重要。第三,如果预期覆盖范围会随着查询序列的变化而变化,例如转录组学,宏基因组学,异质细胞样品或预扩增文库(例如扩增子或单细胞测序),那么大多数k- mer工具(以及相关的自动ķ-mer信任阈值)不合适-例如,使用汉明图和汉明邻域的较新工具除外。相反,大多数MSA工具将适用于此类数据(“删除覆盖范围假设的统一性”一节)。第四,如果您的分析对单核苷酸错误非常敏感(例如,调用SNV时),则您可能希望使用具有更复杂(和/或更具体)错误模型的工具(例如,“利用统计错误进行去噪”部分中的工具楷模',补充说明S5和补充数据),尽管通常会以计算为代价。第五,如果您的数据集不是来自同质的单倍体样本,那么具有重复和/或单倍型模型的工具之一可能会改善错误校正(例如,“重复和单倍型模型”部分中的工具),但也需要更多的计算资源。第六,可扩展性的重要性主要取决于数据集的大小,并且还将影响可以使用的工具类型(就所使用的数据结构而言)。然而,对于所有主要的方法,该方法MSA,基本ķ-mer方法和read后缀方法-最新的工具使用有效的实现,并且都应扩展到大型数据集。因此,最终,工具的选择将取决于每种情况下对错误校正的要求。
表2。
建议考虑使用哪些工具对哪些数据和分析类型进行基准测试
| 标准 | 属性 | 数据集 | 需要考虑的工具 |
|---|---|---|---|
| 平台 | 454 | 任何(均聚物错误!) | HECTOR,KEC,Acacia,AmpliconNoise,DeNoiser,PyroNoise |
| 平台 | 牛津纳米孔 | 任何(错误率很高!) | 纳米芯 |
| 平台 | 太平洋生物 | 任意(错误率高!,偏差小) | proovread,LoRDEC,ECTools,pacbio_qc,PBcR,LSC,AHA |
| 资料属性 | 覆盖范围不均 | 元基因组学,转录组学,全基因组扩增 | 抹子,蓝色,BayesHammer,QuorUM,fermi,Hammer,ALLPATHS-LG,爬行动物 |
| 资料属性 | 主要是替代错误 | 完整的基因组学,Illumina | BFC,打火机,抹子,BayesHammer,QuorUM,Musket,RACER,SGA,SOAPdenovo2,fermi,REDEEM,Hammer,SysCall,DecGPU,ECHO,HiTEC,ALLPATHS-LG,Reptile,CUDA-EC,SOAPdenovo,Quake,SHREC,FreClu, EULER-USR |
| 资料属性 | 插入错误普遍存在 | 任何(尤其是454,Ion Torrent,PacBio) | Fiona,蓝色,SEECER,珊瑚,ShoRAH,Hybrid-SHREC |
| 资料属性 | 许多重复或单倍型 | 元基因组学,复杂的基因组(例如真核生物) | SEECER,Acacia,SGA,SHoRAH,EULER-USR |
| 资料属性 | 两种单倍型 | 二倍体基因组 | 回声 |
| 分析类型 | 对单核苷酸错误敏感 | 例如用于SNV分析 | Fiona,REDEEM,ECHO,SysCall,Quake,FreClu |
工具按发布时间顺序给出,最新的工具优先。建议基于对不同纠错方法的文献综述。
不包括2008年或更早之前的工具。454,Oxford Nanopore和PacBio的特定于平台的工具仅分别在第一,第二和第三行中提到。其他行中列出的工具都应适用于来自不同平台的各种数据类型。
实现所讨论的纠错方法的软件概述(更多信息,请参见 补充表S2)。这包括纠错只是更复杂过程的一个步骤的软件,例如在从头组装或单倍型重建的软件中。Q列指定工具是否使用基本通话质量得分。第一列指定工具是否可以校正indel。值“ 0.5”表示可能存在indel校正的可能性,即工具已经隐式校正了部分但不是全部indel,或者可以轻松扩展以对其进行校正。
| 工具 | 主要方法 | 数据结构) | k聚体长度 | k选择推理 | 全球k- mer信任阈值 | 错误模型 | 更正 | 问 | 一世 | 进一步的区别 |
|---|---|---|---|---|---|---|---|---|---|---|
| 金合欢 | MSA和集群 | RLE前缀的哈希 | 6(RLE!) | – | Emp Hopo UC和OC概率 | 迭代MSA,使用统计测试的迭代聚类精炼,聚类缺点 | – | 0.5 | RLE的6聚体读取前缀;使用3个bin测试所有RL差异:uc,main mode,oc;打破与一致的不匹配的缺点; 包括解复用 | |
| AHA | 读取对齐 | 支架中的隐式LR corr | – | 0.5 | LRs从SRs现有的重叠群的脚手架,通过短读对LRs的初始corr | |||||
| 全路径 | k聚体 | – | 16、20和24 | – | Dyn:emp k -mer cov dist的第一位置 | – | 每次读取的最小汉明D(Q加权)校正 | 1个 | – | 仅当三个不同ks的所有k -mers均固体时, 读取正确 |
| 全路径 | ķ -mer频率 | – | 24 | 平衡uniq与灵敏度 | – | 多数赞成票(Q加权) | 1个 | – | 24-mer,中间有1个碱基缺口(每13个碱基对应),连续的24-mer:所有cols具有> 6个读数,对读数中的变化进行一致性检查 | |
| 扩增子噪声 | 流程图和Corr读取提示(EM) | – | (i)Emp pyroseq错误,(ii)emp PCR错误(学习的混淆矩阵) | (i)和(ii):混合模型的EM,用于假设某些生成序列的obs freq的流程图/读取生成可能性 | – | 1个 | 每个真实序列具有1个指数dist分量的混合模型,可从校正的pyroseq读数中获得PCR误差corr的混淆矩阵,目标是454个扩增子的焦磷酸测序 | |||
| 蛛网 | MSA | – | 8-24 | – | 多数col投票(Q加权),仅在alt基数的cov差异较大时适用 | 1个 | 1个 | 嵌合体检测,如果中心区域的cov降低(=与其他读段的重叠下降) | ||
| 自动编辑 | MSA和色谱图 | – | 各区域色谱峰的分辨度 | 使用来自装配/映射的基本频率进行基本召回 | 1个 | 1个 | 基础调用,针对Sanger数据的色谱图上下文信息集成 | |||
| 贝叶斯·汉默 | k- mer规格和汉明图 | 重复排序的k- mer列表(Hamming图),不交集(连接的组件) | 21(防御) | – | 误差概率= Q | 通过对连接的组件进行子集群和基于读取的校正来改善Hammer | 1个 | – | 贝叶斯信息准则对连接组件的k均值聚类进行惩罚;基于簇质量的k聚体谱;输出读取 | |
| BFC | k聚体 | 布隆过滤器和哈希表 | 31,55 | 权衡重复分辨率与k- mer覆盖率 | 男子:3 | – | 从最长的受信任区域中进行遍历读取,并通过扩展对Corr的惩罚找到最佳校正 | 1个 | – | 从费米精制而成;彻底搜索corr,包括可信任的k- mers的corr ;跳过可信赖的k- mers中 高质量碱基替代品的测试 |
| 保佑 | k聚体 | 哈希表(计数),bloom过滤器(k- mer规格) | – | (i)#k -mers / 4 ^ k≤0.0001(ii)最大#corr基数 | Dyn:emp k -mer cov dist的第一位置 | – | 在读取中的固体k- mer孤岛之间找到固定的最小编辑D路径,超出读取末端的路径会产生末端错误 | 1个 | .5 | 逆转某些布隆过滤器误报更改,通过读取扩展更好地纠正读取端的错误 |
| 布卢库 | k聚体 | 布隆过滤器 | 31(防御) | – | 人(def = 3–6) | – | 作为步枪 | – | .5 | 作为步枪;仅接受由多个实体k- mers支持的corr,以避免Bloom假pos corr |
| 蓝色 | k聚体 | 哈希表(已分区) | k > 20(手动) | 优衣库(手动) | Dyn每次读取thr:k -mer cov的谐波平均值的1/3 ,不包括低cov k -mers | 低COV,COV下降,跳跃运行结束 | 所有可行k- mer corr 的从左到右的深度优先遍历 | – | 1个 | 将k -mer规范生成与错误corr解耦,从而允许数据集之间进行互相关 |
| 珊瑚 | MSA | 哈希表(k -mer读取地图),邻接表 | k≈log_4 | G | | Uniq,但k <L / 2(读取中存在正确的k- mer) | – | Q加权rel基Covs的Corr w / thr | 1个 | 1个 | 考虑MSA Q | |
| CUDA-EC | k聚体 | 计数布隆过滤器 | 20 | 'Illumina def' | 男人:假定真k -mers的泊松分布 | – | 最小Hamming D corr,最大#k -mer corr首先,每次读取都贪婪 | 1个 | – | Bloom过滤器介绍和CUDA并行化 |
| DecGPU | k聚体 | 布隆过滤器 | 21(防御) | – | 人(def = 6) | – | 多数赞成票 | – | – | CUDA和MPI并行化 |
| 去噪器 | 流程图类(EM) | – | (AmpliconNoise:(i)) | (AmpliconNoise:(i)) | – | 1个 | 使用精确的前缀进行快速聚类,目的是对454个扩增子进行焦磷酸测序 | |||
| 回声 | MSA | 哈希表(k -mer读取地图),邻接表 | k≈L / 6 | 权衡讨论 | 读取pos特定的混淆矩阵(估计为EM) | 在混淆矩阵加权的col处给定基础covs的基础调用的最大后验估计 | – | – | 杂合度模型 | |
| ECTools | MSA | – | – | LR的Corr与来自预组装SR的单元 | – | 1个 | (i)来自特别代表的单位;(ii)使LR与单位对齐;(iii)使用最长的递增子序列进行优化;(iv)纠正单位 | |||
| 雷达 | 读位置的k -mer规格和k -mer频率群 | – | – | – | Dyn:emp k -mer cov dist的第一位置 | – | 删除错误基础(拆分读取) | – | 0.5 | GC含量调整(每k- mer),在读取低k- mer频率(可变带宽均值漂移方法) 内,识别并除去作为pos簇的错误基础 |
| 欧拉 | k聚体 | – | 20、100 | – | 男人:X | – | 最小Hamming D corr,最大#k -mer corr首先,每次读取都贪婪 | – | – | k -mer规范的初始用法用于错误更正 |
| 欧拉 | k聚体 | – | 15–20 | 较短的k- mers始终稳定 | 男人:X | – | 每次读取的最小Levenshtein D corr,dyn编程 | – | 1个 | |
| EULER-SR | k聚体 | – | 15 | – | 男人:假定真k -mers的泊松分布 | – | 最小Hamming D corr,最大#k -mer corr首先,每次读取都贪婪 | – | – | k- mer信任thr的迭代松弛(高cov至低) |
| EULER-USR | k聚体 | 重复读取前缀的图形(简体de Bruijn) | 20 | – | Dyn:泊松和高斯混合拟合的第一位置 | – | 最小汉明D corr,最大#k -mer corr首先,贪婪的每个readprefix,后缀corr w /重复前缀图 | – | – | |
| 费米 | k- mer规格和k- mer频率 | 后缀数组(BWT和FMD索引),哈希表(k -mer spec) | 23(防御) | – | 男人:def = 3 | – | 前缀多数票支持低Q基本OCC | 1个 | – | |
| 菲奥娜 | ķ -mer频率 | 后缀数组(部分) | 各种 | 作为HiTEC | Dyn:从k和错误概率 | 层次统计模型 | 每个可能的错误:正确阅读的所有重叠pos的多数表决,读内的diff corr pos之间的贪婪 | – | 1个 | 统计模型的详细信息:请参阅 补充资料 |
| FreClu | 丛书读着汉明D | 哈希表,表示已排序的读取列表(Hamming D) | 读取pos和Q特定的碱基混淆矩阵 | 伯爵 | 1个 | – | 按汉明距离进行聚类读取,从covs创建聚类树,然后仅映射根读取,扩展POLYBAYES错误模型,针对RNAseq | |||
| 锤子 | k- mer频率和汉明图 | 重复排序的k- mer列表(Hamming图),联合查找(连接的组件) | 55(防御) | 权衡讨论 | – | 使k- mers的Corr最大化关联组件的最大似然缺点,并通过对Q加权k - mer covs 进行多数表决来打破似然关系 | 1个 | – | 没有统一的cov假设,针对最小cov(def = 1)的singletonCutoff,如果cov高于,则saveCutoff可在每个集群中保持多个k -mers,仅输出k -mers | |
| 赫克托 | k-hopo规格 | 布隆过滤器和哈希表 | 21(def,k-hopo) | – | Dyn:emp k -mer cov dist的第一位置 | – | 作为步枪,但使用k-hopos来测试基本信任 | – | 1个 | K-hopos代替ķ聚体为EMP COV DIST:K-HOPO编码K内由1字节的连续RLE HOPO运行; 最多允许3个碱基插入 |
| 海泰克 | ķ -mer频率 | 后缀数组 | 各种 | 给定每个基准错误率的最小假阴性或假阳性 | Dyn:从| G |,L,N,替换错误概率 | – | 明确的正确性:多数票,提前2 nt打破平局 | – | – | 迭代各种k- mer大小:值要么最小化#个不可纠正的读数(假阴性),要么最小化#个可破坏的读数(假阳性) |
| 混合SHREC | ķ -mer频率 | 后缀特里 | (SHREC) | (SHREC) | 通过调整参数alpha调整为当前k | – | (SHREC)加上向上和向下查找一个Trie级别(为indel corr扎根) | – | 1个 | 检查多个k |
| 韩国电子学会 | 读pos和MSA的k- mer规格和k- mer频率群 | 杂凑 | 25(防御) | 清除弱/固体分离与错误解决方案 | Dyn:在emp k中的0 cov的第一个拉伸结尾-mer cov dist | – | 弱区域中每次读取的最小编辑量,产生的最大k -mer cov | – | 1个 | 识别错误区域,如在EDAR中一样;遍历错误更正几轮;从独特读段的MSA中消除非常低频的单倍型;未针对其他k- mer corr进行 基准测试 |
| 打火机 | k聚体 | 布隆过滤器 | 23 | 正确基准的最大增益 | – | 像BLESS | 1个 | – | 三遍读取:(i)将k -mers子采样到第一个Bloom过滤器中;(ii)长度为k的受信任读取部分到第二Bloom过滤器中;(iii)为BLESS | |
| 洛雷德 | k聚体 | 布隆过滤器中的de Bruijn图 | 17-21(def) | – | 人(def = 2–3) | – | 遍历SR de Bruijn图的弱LR区的Corr | – | 1个 | SR的实心k- mers的de Bruijn图; 实心k聚体区域作为种子通过图遍历实现弱LR区域的校正 |
| LSC | MSA | – | RLE压缩读取的混合读取集一致性 | – | 1个 | (i)RLE压缩SR和LR,(ii)将RLE SR映射到RLE LR,(iii)将LR校正为MSA利弊并修整为SR覆盖的区域 | ||||
| 错误的 | MSA | 表(k -mers读取图) | – | 如果col未定义核苷酸pos,则进行多数表决(检验偏差一致的概率模型) | 1个 | – | 读取中的Alt碱基连锁(差异重复/多态性与错误) | |||
| 滑膛枪 | k聚体 | 将过滤器放入哈希表 | 21(防御) | – | Dyn:emp k -mer cov dist的第一位置 | – | 双面保守,一面进取和基于投票的改进 | – | – | 如果更改使最左和最右重叠的k -mer保持一致,则会产生明确的错误,否则,从实心区域扩展和col多数投票加上前瞻性k -mer一致性检查(def = 2)加上每k -mer的最大更改(def = 4) |
| 杂种 | MSA | 后缀数组 | k≈log_4(200 * | G |) | (地震) | Dyn:给定错误概率的exp k -mer occs的 一半 | – | 多数赞成票 | – | 1个 | MSA种子的K- mer Cov thr |
| N-corr | 454中的Hopo模式读取 | – | Hopo模式 | – | – | – | ||||
| 纳米芯 | MSA | – | – | SR对齐的缺点 | – | 1个 | 将SR与LR对齐,动态找到最佳对齐方式 | |||
| pacbio_qc | 读取过滤器(SVM) | 请参阅LIBSVM软件包 | 平均Q和CCS通过读取次数 | 支持向量机回归训练在已知的尖峰 | 1个 | |||||
| PBCR | MSA | – | 混合读集Corr | – | 0.5 | (i)将SR映射到LR,(ii)将LR正确化为MSA缺点 | ||||
| 盆模型 | 朗读汉明D | – | 误差概率= Q | 用群聚法估计汉明邻域的最大似然波茨模型 | 1个 | – | ||||
| 总理 | k- mer频率(HMM,EM) | – | 优化性能 | Uniq与足够的cov | 从EM读取的pos,Q,cov和碱基混淆矩阵的k聚体跃迁概率 | 带HMM的最大序列产生可能性 | 1个 | – | ||
| 证明 | 读取映射 | 每个LR的比对矩阵 | 带有惩罚错误概率的惩罚映射 | 以多数票召集的缺点 | 1个 | 映射的处罚反映了PacBio错误概率;SR基础支持的质量得分;嵌合体检测;w /增加灵敏度的迭代 | ||||
| PSAEC | ķ -mer频率 | 后缀数组(部分) | 20(前) | – | Dyn:从| G |,L,N,替换错误概率 | – | (HiTEC) | – | – | 仅使用部分后缀数组进行运行时和内存优化 |
| 火焰噪声 | 流程图集群 | Emp pyroseq错误 | 假设一定的生成序列,混合模型的电磁场流程图对obs freq生成可能性的影响 | |||||||
| 雷神之锤 | k聚体 | 位数组索引(k -mer规范) | k≈log_4(200 * | G |) | 对于k -mer空间4 ^ k / 2中的k -mer occ, Prob = 0.01 | Dyn:混合dist模型拟合和似然比 | Q特定混淆矩阵(从明确的初始校正中学习) | 最大似然k -mer变化,最小Q罐误差pos 1st | 1个 | – | 第一个关于k -mer长度折衷的正确讨论,复杂的k -mer dist混合模型:弱=伽马,实心=泊松,重复k -mer多重性= Zeta |
| 法定人数 | ķ -mer频率 | – | 24(防御) | – | – | 修剪是否没有带有Cov的替代碱基,如果只有一个带有COV的替代碱基,则更正,如果多个带有非零COV的替代,则进行单向超前,通过冠状连续性检查来打破联系 | 1个 | – | 没有统一的cov假设:在读取时突然cov下降时校正k -mers,可以去除污染物k -mers,进行修整,保守,不检查高cov k -mers | |
| 赛车手 | k聚体 | 哈希表 | “根据| G |计算” | – | Dyn:来自| G | | – | 明确无误到可靠 | – | – | |
| 叙事 | 读取计数(EM) | – | 对齐列中Q值的平均值给出特定于读取和pos的错误概率 | 伯爵 | 1个 | EM的exp读取计数 | ||||
| 赎回 | k聚体 | 稀疏 | 11(防御) | 非重复k -mers的 Uniq | 人(def = 20),但dyn频率计数 | 学习稀疏的k- mer混淆矩阵 | 将所有覆盖的k -mers上的每个读取pos设置为最大概率的nt ,弱k -mers 的读取的corr | – | – | 计算给定obs k -mer频率的exp k -mer频率(使用k -mer误读概率的最大似然EM ) |
| 爬虫 | k- mer频率和汉明图 | 重复排序的k- mer列表(Hamming图) | k≈log_4 | G | 或10≤k≤16 | 平均K均值 | 男人:高和中置信度k -mer thrs(Q) | – | 如果瓦片具有低cov和低Q pos,则每次连续两个k- mers(瓦片)的 最小汉明D corr | 1个 | – | 适用于校正罐的k -mer频率比率thr与校正步骤中校正罐的k -mers(def = 2) |
| SEECER | MSA,col clust和HMM | 哈希表(k -mers读取映射) | 17(防御) | 了解每个MSA重叠群的HMM参数 | 每个重叠群一个HMM,从Viterbi的算法通过对数似然读取对HMM的分配 | – | 1个 | 针对RNAseq数据,通过不匹配cols上 的k均值的谱簇和谱弛豫将多态性与错误分离 | ||
| SGA | ķ -mer频率 | 后缀数组(BWT和FM索引) | 31(防御) | 从具有各种('sga stats')的读取子集的错误Corr中选择Emp | Dyn:emp k -mer dist的 loc min | – | 最左和最右重叠k -mer检查thr高于thr的alt碱基 | 1个 | – | 可选仅将PHRED Q的基数pos用作k- mer计数和thr确定 |
| SGA | MSA | 后缀数组(BWT和FM索引) | – | 如果单个基数高于corr cov thr(def = 3),则Corr冲突cols | 1个 | – | 检查不匹配链接(read的冲突col =冲突cov thr之上的多个alt基;从read的corr MSA中排除所有冲突均具有不匹配的read) | |||
| ShoRAH | MSA和阅读提示 | 哈希表 | – | 单倍型内的位置多数规则(在每个seq pos重叠的三个窗口中) | – | 1个 | Clust使用Dirichlet过程混合物后部距离的Gibbs采样器读取单倍型 | |||
| SHREC | ķ -mer频率 | 后缀特里 | [min {log_4(| G |),log_4(n)} + q]≤k≤s | n = #reads,q:(1/4)^ q <p,s:特里水平的低cov thr | 通过调整参数x调整为当前k | – | 如果实际节点的cov低于exp节点的cov(±SD *调整参数),则重分支当前Trie级别 | – | – | 检查多个k |
| SleepEC | 读取频率和汉明图 | 节点列表,节点属性,节点链接 | 读取长度 | 读取pos特定的基础混淆矩阵 | 实际阅读量与预测值之间的统计检验 | – | – | 对于RNAseq读取;建立汉明图,从受信任的连接组件中学习基本替换矩阵 | ||
| SOAPdenovo | k聚体 | 哈希表(k -mer freq) | k≈log_4 | G | | 平均K均值 | 人 | – | Min Hamming D corr通过扩展高cov区域,多数col投票,dyn编程来实现每次读取 | – | – | |
| SOAPdenovo2 | k聚体 | 哈希表或索引表 | k≈log_4(20 * | G |) | k- mer空间> 10x基因组k- mer空间 | 人 | – | 上校对明确的错误进行投票,对可能的变更路径(植根于正确的位置)进行投票,以解决不确定的错误 | – | – | |
| SysCall | MSA分类器(逻辑回归) | 矩阵(行=锅池pos,列=特征) | 后验概率基于(i)与邻居的Q差异,(ii)序列上下文,(iii)链偏向 | Logistic回归模型,将每个锅位的误差从误差中区分出来(thr 0.5) | 1个 | – | ||||
| 镘 | k聚体 | 哈希表(以k -mer作为键,以corr pos的最大Q作为条目) | 作为地震 | 作为地震 | – | 误差概率= Q | 比较Musket:(i)间隙k -mer(砖)校正,(ii)单面k -mer校正,用于读取末端和连续的错误pos | 1个 | – | k -mer spec完全基于Q(全局Q thr),通过基于读取的低Q pos增强来迭代扩展 |
缩写:#=数量,alt =替代项,alg =算法,BWT = Burrows Wheeler变换,CCS =圆形共识序列,clust =聚类,col =列,cons =共识,corr =校正,cov =覆盖率, D =距离,def =默认值,diff =差异,dist =分布,dyn =动态,EM =期望最大化,emp =经验,exp =期望,FM = Ferragina Manzini,freq =频率,| G | =基因组大小,HMM =隐马尔可夫模型,Hopo =均聚物,I =插入缺失=插入/缺失,k = k-mer长度,L =读取长度,LR =长时间读取,loc =本地,man =手动,max =最大值,min =最小值,MSA =多序列比对,N =#reads,na =不可用,obs =已观察到, oc =超越,occ =出现,pos =位置,底池=电位,概率=概率,Q =质量,rel =相对,repl =复制,RLE =游程编码,spec =频谱,SR =短读,SVM =支持向量机,thr =阈值,uc =通话,uniq =唯一性,X =经验。
纠错工具的引用和软件URL
通常,这里介绍的有关错误,它们的偏差,覆盖率偏差及其对特定测序平台的依赖性的知识只有很少的工具考虑在内。但是,更具体的方法将提高纠错的准确性,从而改善下游分析:读取映射和从头开始程序集在计算上将变得更易于处理,并且会提供更好的结果,从此类比对派生的变体调用将更加准确。但是,更高的特异性可能会将工具的范围限制为某个平台或某种数据集。相反,考虑到我们刚才讨论的六个方面,我们认为未来的工作应该针对整个错误纠正工具包,该工具包应免费提供给任何使用者和开源,以鼓励分布式开发,例如针对由Genome Analysis Toolkit(GATK; [ 114 ])进行变体发现,或由SeqAn [ 115 ]进行测序数据分析],这是一个基于纠错器Fiona的库工具包。根据手头的数据集,这样的工具箱应该最佳地能够选择地考虑indel,从而做出选择是假设覆盖均匀以减少计算量,还是避免这种假设,从而选择特定于平台的错误模型从(甚至从数据集中学习模型的参数),以允许使用有关预期单倍型的先验信息,并在可能的情况下重复查询序列的组成,或者能够从数据本身推断单倍型和重复,并在使用三种主要方法中的一种时应具有灵活性,这取决于对特定数据集的要求组合在计算上最有效的方法。这表明采用模块化方法进行纠错,
我们相信,此处给出的错误纠正方法和工具的概述和比较将为用户和开发人员提供信息,并使社区能够:将来继续进行提议的错误纠正软件的模块化开发,以灵活地使用这种模块化方法结合来自不同错误纠正方法的想法(如PacBio,Oxford Nanopore和454错误纠正最近开始使用),以利用有关错误及其在该领域及以后发展中的偏见的更多知识(例如SNV调用),并在针对许多不同数据类型和分析设置的全面而有代表性的数据集上,为此处列出的工具创建有意义的比较基准。
-
每个测序平台都有一个与各自的测序技术相关的独特错误概况。这包括在除PacBio之外的所有平台上对极端GC含量的充分掩盖,其中离子洪流表现出最大的偏见。关于单核苷酸错误,对于所有其他平台,Illumina和Complete Genomics显示的取代错误比插入缺失错误更多。总的来说,Illumina的短读错误率最低,而PacBio和Oxford的长单分子测序读错率最高。但是,PacBio错误似乎是真正随机的,而Illumina错误尤其表现出特定的偏差,例如由于某些序列基序。
-
旨在促进原始读取数据中的错误校正的最早方法是基于k- mer谱,其目的是促进它们的从头组装。这个想法是根据读取的数据创建一个受信任的k个长度序列(k个聚体)的列表,并朝该频谱校正所有不信任的k个聚体,而信任一个k个聚体通常需要一定的最小覆盖范围。该方法需要确定的最重要参数是k-mer长度和覆盖范围阈值。对于这两者,没有选择在所有情况下都效果最佳,并且较新的工具会尝试完全避免将全局覆盖阈值应用于在整个查询序列中覆盖范围不均匀的数据。最近,通过使用布隆过滤器来存储k- mer光谱和相关信息,已经实现了重大的性能改进。
-
与从头组装紧密相关的另一种早期纠错方法是生成所有读取的MSA并进行纠正以达成共识。但是,最初的“全部对所有”比对方法变得难以用于下一代高通量测序的读取,并且仅在以后使用可能的读取重叠的智能索引进行适应以播种比对(例如,使用k- mer索引或读取聚类)技术)。通常,基于MSA的方法通常非常适合解决indel错误,不需要全局频率阈值或选择k-mer长度,并且可以轻松地将读取的上下文信息整合到更正决策中(例如重复消除歧义或单倍型连锁)。最近,随着单分子测序技术的出现,它们产生了越来越少的读取次数,但读取时间却越来越长,它们变得越来越流行。
-
第三种主要方法是使用后缀数组(或其衍生物)进行纠错。数据集中所有读取的所有可能后缀的排序列表不仅可以用于播种MSA,而且还可以有效地查询自身,以提供任何可能长度的k- mers的覆盖率值。实际上,这产生了k- mer频谱方法的一种更灵活的版本,并且最近使用BWT和FM索引的实现方式使该方法可用于高吞吐量数据集。值得注意的是,相应的工具是最早在测序过程中采用错误统计模型并自动选择和检查多个k- mer长度以决定错误的工具之一。
-
在过去的几年中,错误和重复模型已经有了很大的发展。较早的工具只能以统一的错误率工作,而较新的工具通常使用混淆矩阵来替代错误,这可能取决于质量得分和读取位置,并且某些工具使用单独的插入和删除率。这样的值对于平台是已知的,或者可以从所考虑的各个数据集合中近似得出。而且,工具已经开始以概率方式显式地对测序过程进行建模。同时,模型试图通过查询特定的数据结构来解释所查询序列中的重复或单倍型。通过查看读段中的变异链接;或通过概率估计单倍型。
-
到目前为止,只有两项研究提供了此处介绍的工具的子集的基准。结果,大多数错误校正器的用例不允许在不执行特定基准的情况下进行明智的工具选择,尤其是汇编程序的最终用户大多依赖于其所选软件中内置的错误校正器。高通量测序社区将从全面数据集的特定比较基准中受益匪浅。这些应该代表不同的数据类型和用例。此处提供的方法概述可以指导选择工具以针对特定设置进行基准测试。
致谢
作者要感谢Andreas Bremges,Ivan Gregor,Andreas Kloetgen和Niranjan Nagarajan对手稿的宝贵反馈。
资金
这项工作得到了德国杜塞尔多夫海因里希海涅大学和亥姆霍兹学会的支持。