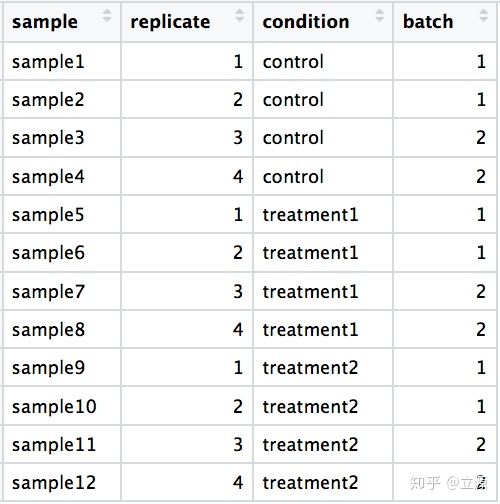
実験計画で考慮すべき要素
RNAの抽出方法とRNA-Seqライブラリー構築の実験手順を理解することは、RNA-Seq実験の設計に非常に役立ちますが、差次的発現分析の品質に深刻な影響を与える可能性のあるいくつかの特別な要因を考慮する必要があります。
これらの重要な考慮事項は次のとおりです。
- レプリケートの数とタイプ
- 混乱を避ける(交絡)
- バッチ効果の処理
それぞれの考慮事項を詳細に検討し、ベストプラクティスと最適化された設計について説明します
複製
実験の繰り返しには、技術的複製と生物学的複製が含まれます
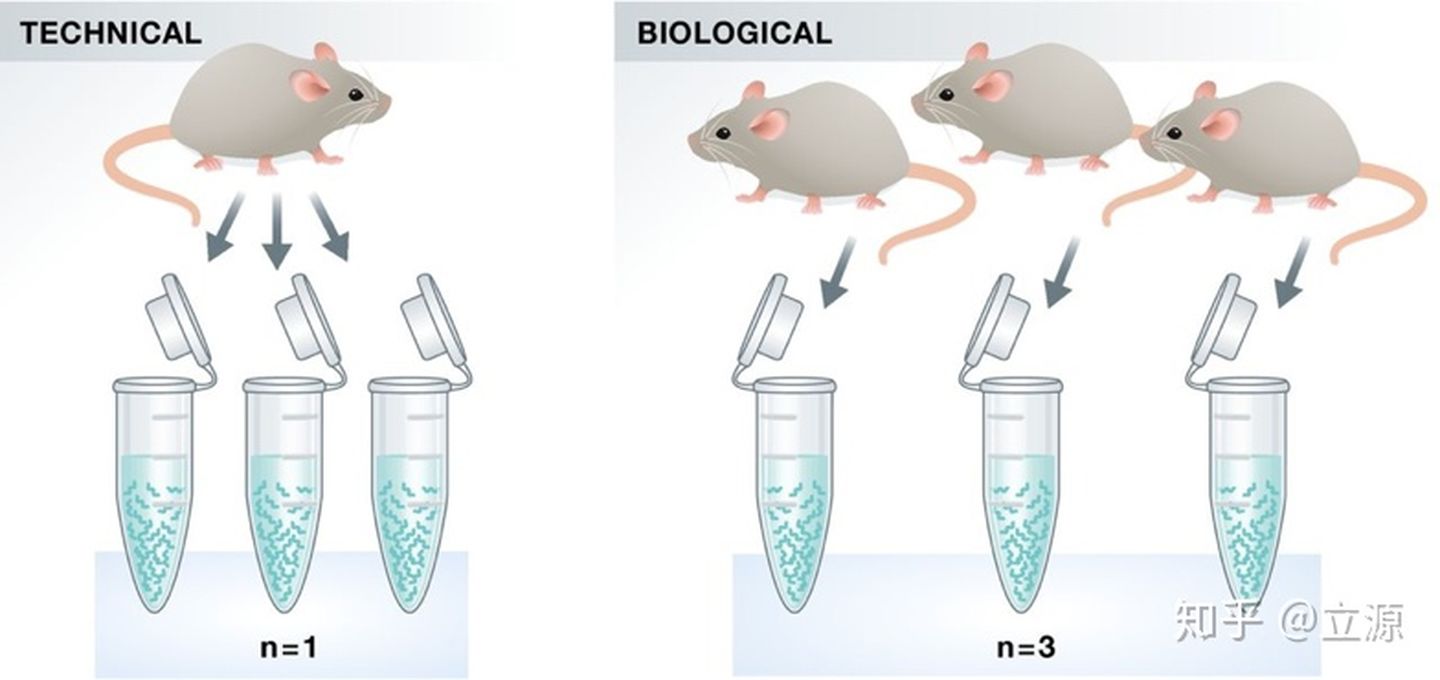
画像ソース: Klaus B.、EMBO J(2015)34:2727-2730
- 技術的な繰り返し:同じ生物学的サンプルを使用して技術的または実験的な手順を繰り返し、技術的な違いを正確に測定し、分析でそれらを削除します
- 生物学的複製:同じ条件下で異なる生物学的サンプルを使用して、サンプル間の生物学的差異を測定する
マイクロアレイの時代では、技術的な繰り返しが必要であると考えられていますが、現在のRNA-Seq技術では、技術的な変化は生物学的変化よりもはるかに小さいため、技術的な繰り返しは必要ありません。
逆に、差次的発現解析では、生物学的重複が絶対に必要です。マウスやラットの場合、異なる生物学的サンプルの原因を判断するのは簡単かもしれませんが、細胞株でそのような判断を下すのははるかに困難です。この記事は、細胞株の複製についていくつかの非常に良い提案をします。
差次的発現分析の場合、生物学的複製が多いほど、生物学的変異の推定が向上し、平均発現レベルの推定がより正確になります。これにより、データモデリングがより正確になり、より差異のある遺伝子が特定されます。
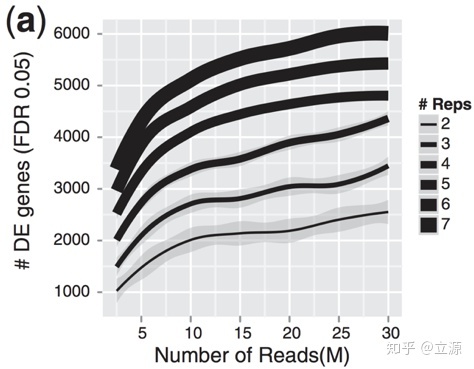
画像ソース: Liu、Y.、et al。、Bioinformatics(2014)30(3):301–304
上の図に示すように、生物学的反復は、シーケンス深度、つまり各サンプルで測定された読み取りの総数よりも重要です。この写真は、検出された差次的遺伝子の数におけるシーケンス深度と生物学的リピートの数との関係を示しています。生物学的反復の増加に伴い、シーケンシングの深さが増し、より差異のある遺伝子が見つかることは注目に値します。したがって、より多くのリピートは一般に、より高いシーケンシング深度よりも優れており、より高いシーケンス深度は、発現の少ない差次的遺伝子を検出し、アイソフォームレベルの遺伝子発現を実行するためにのみ必要です。
混合サンプル(サンプルプーリング) :可能であれば、個々の/実験の混合は避けてください。ただし、どうしても必要な場合は、各サンプルセットの混合を1 回の繰り返し(SINGLEレプリケート) として扱う必要があります 。レプリケート間で同程度の変動を確保するには、混合されたサンプルセットごとに同じ数の個体を混合します。
たとえば、コントロールの繰り返しに十分なデータを取得するために少なくとも3人、治療の繰り返しに十分なデータを取得するために少なくとも5人が必要な場合、コントロール用に5人、治療条件用に5人を収集する必要があります。また、同じ条件下で収集された個人が性別、年齢などで類似していることを確認します。
バルクRNA-Seqの場合、ほとんどの場合、重複はより高いシーケンス深度よりも優先されます。ただし、ガイドラインは、実行した実験と必要な分析によって異なります。繰り返しとシーケンス深度の実験計画を支援するためのいくつかの一般的なガイドラインを以下に示します。
- 一般的に使用される遺伝子レベルの差次的発現:
- ENCODEガイドラインでは、サンプルごとに30,000,000リードのシングルエンドシーケンスを推奨しています
- 十分に繰り返す(> 3)場合、通常、サンプルあたり15,000,000回の読み取りで十分です。
- 可能であれば、より多くの生物学的複製にお金を使う
- 一般的に推奨される読み取り長> = 50 bp
- 低発現遺伝子の遺伝子レベルの発現差の検出
- 同様に、複数の複製は、シーケンス深度を増やすよりも優れています
- 発現レベルによると、ディープシーケンスは少なくとも30〜60,000,000回以上の読み取りです(十分な繰り返しがある場合は、30,000,000から開始します)
- 一般的に推奨される読み取り長> = 50 bp
- サブタイプレベルでの差次的発現:
- おなじみのサブタイプの中で、サンプルあたり少なくとも30,000,000回の読み取りの深さのペアエンドシーケンスが推奨されます
- 不明なサブタイプには、より深い深度が必要です(サンプルあたり> 60,000,000回の読み取り)
- ペアエンド/ディープシーケンスの代わりに生物学的複製を選択する
- 一般的に推奨される読み取り長> = 50 bpですが、読み取りはエクソンジャンクションを通過する可能性が高いため、結果が長いほど良いでしょう。
- RNA品質の注意深い品質管理を実行します。高RINサンプルを取得するための高品質のライブラリ構築方法と厳密な分析の使用に注意してください
- 他のタイプのRNA分析(イントロン保持、small RNA-Seqなど):
- さまざまな分析によると、さまざまな推奨事項
- 基本的に、生物学的反復が多いほど常に優れています!
注: ゲノムシーケンシングの深さを推定するために使用される要素は「カバレッジ」です。つまり、テストされたヌクレオチドの数がゲノムを「カバー」する回数です。この指標はゲノム(全ゲノムシーケンス)に対しては正確ではありませんが、十分に優れており、広く使用されています。ただし、ゲノムの何パーセントが転写活性であるかを知っていても、遺伝子発現は大きく変動するため、この指標はトランスクリプトームには適用できません。
交絡
紛らわしいRNA-Seq実験は、実験データの2つの異なる変動源の独立した影響を区別できないことを意味します。
たとえば、性別が遺伝子発現に大きな影響を与えることはわかっています。対照群のすべてのマウスが雌で、治療群のマウスが雄の場合、治療効果は性別によって混乱します。治療の効果と性別の効果を区別することはできません。
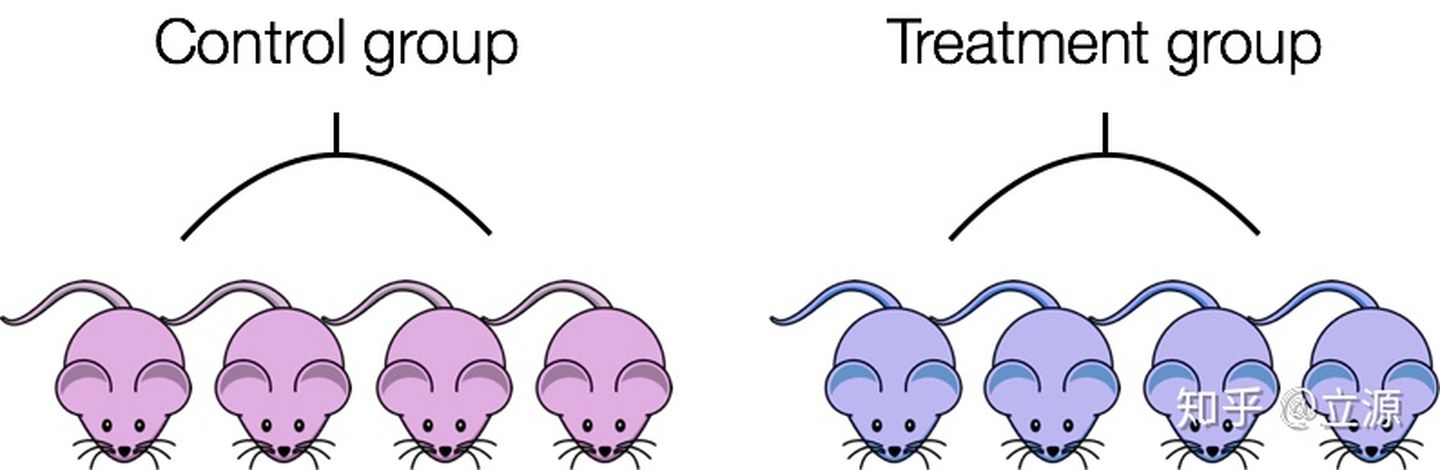
混乱を避けるために:
- 可能であれば、各条件下の動物が同じ性別、年齢、バッチを持っていることを確認してください
- それが不可能な場合は、異なる条件で動物を均等に分割するようにしてください
バッチ効果
バッチ効果は、RNA-Seq分析における重要な問題です。Hicks SC、et al。、bioRxiv(2015)から撮影した写真は、この現象をよく説明しています。実験計画は、各バッチに2つのサンプルグループからのサンプルを含めることで左側に示され、バッチの適切な使用法を示しています。右端には、PCAの例が描かれており、サンプルはバッチに分けられます。これは、遺伝子発現に対するバッチの影響が通常、実験変数からの影響よりも大きいことを示しています。したがって、実験を設計するときは、統計モデルでこれを考慮に入れる必要があります。この問題については、以下で詳しく説明します。
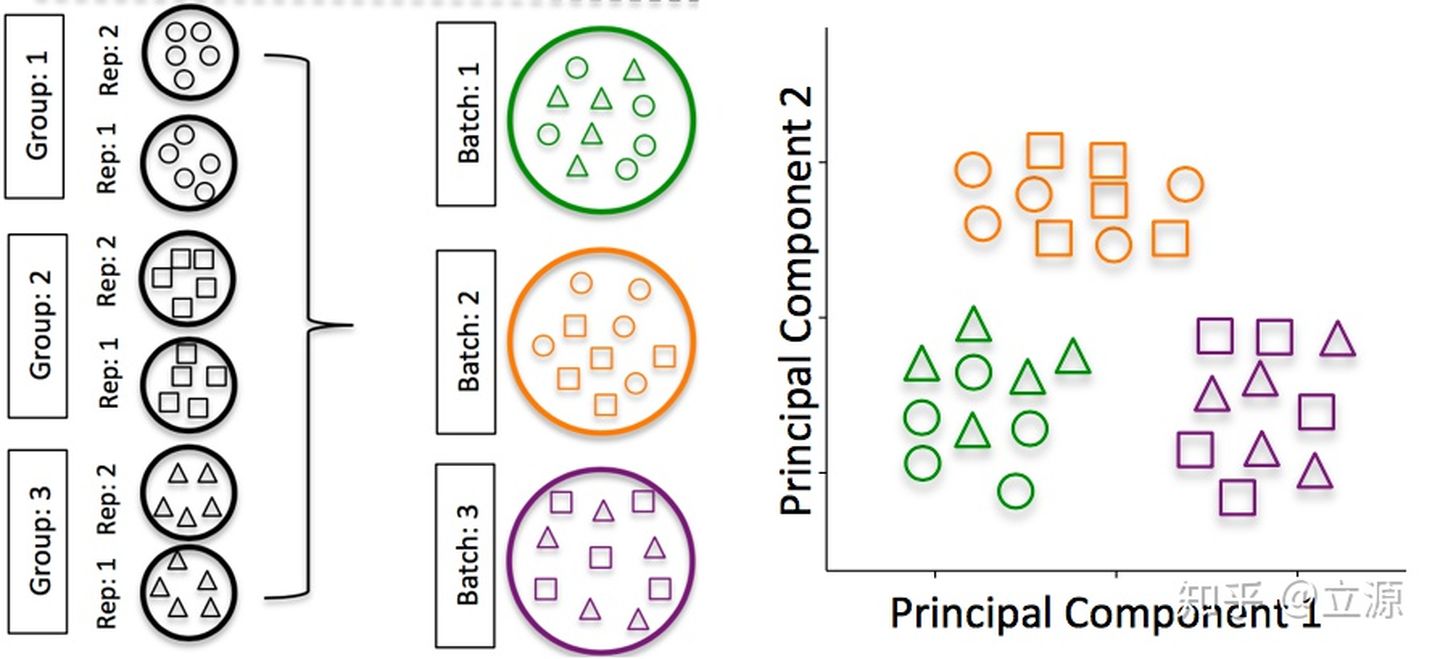
画像ソース: Hicks SC、et al。、bioRxiv(2015)
これらの研究デザインにおける不適切なバッチ処理によって引き起こされる問題は、この記事で詳しく説明されています。
バッチの問題があるかどうかを知る方法は?
- すべてのRNA抽出は同じ日に実行されますか?
- すべてのデータベース構築作業は同じ日に実行されますか?
- すべてのサンプルのRNA抽出またはライブラリー構築は同じ人によって行われますか?
- すべてのサンプルに同じ試薬を使用しましたか?
- 同じ場所でRNA抽出またはライブラリー構築を行ったことがありますか?
これらの答えの1つが「いいえ」の場合、バッチ質問があります。
バッチ問題のベストプラクティス
- 実験計画では、バッチをできるだけ避けてください
- バッチを回避できない場合:
- 実験の異なるバッチを混同しないでください。つまり、1つの条件下でサンプルの1つのバッチを実行するだけではありません。
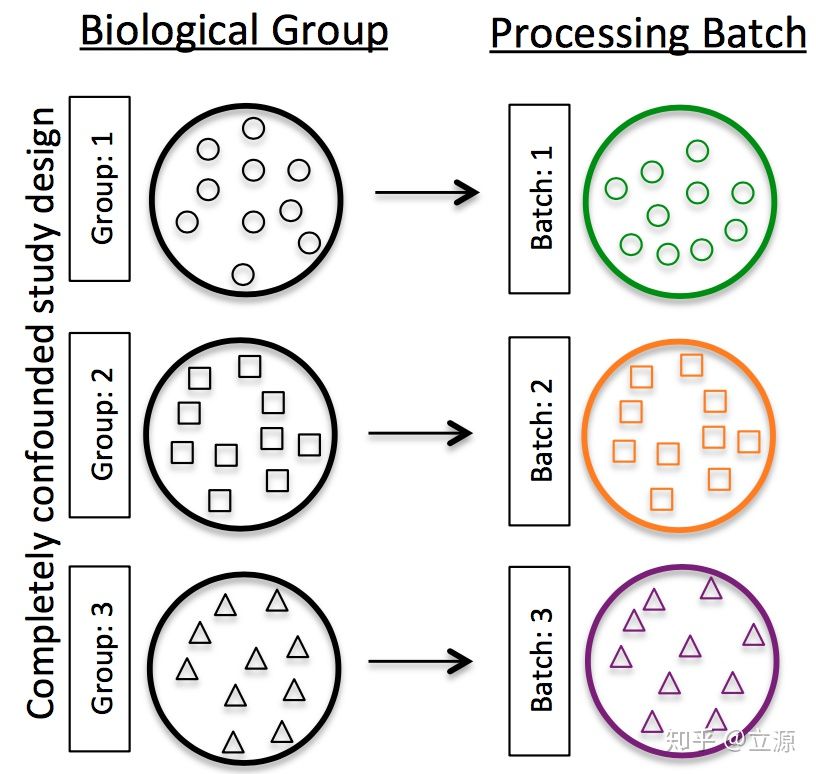
-
- さまざまな種類のサンプルをさまざまなバッチで複製してください。異なる治療条件下で異なる遺伝子を見つけたい場合、またはグループレベルで結論を導き出したい場合は、繰り返しが多いほど良いです(もちろん、2つ以上)。
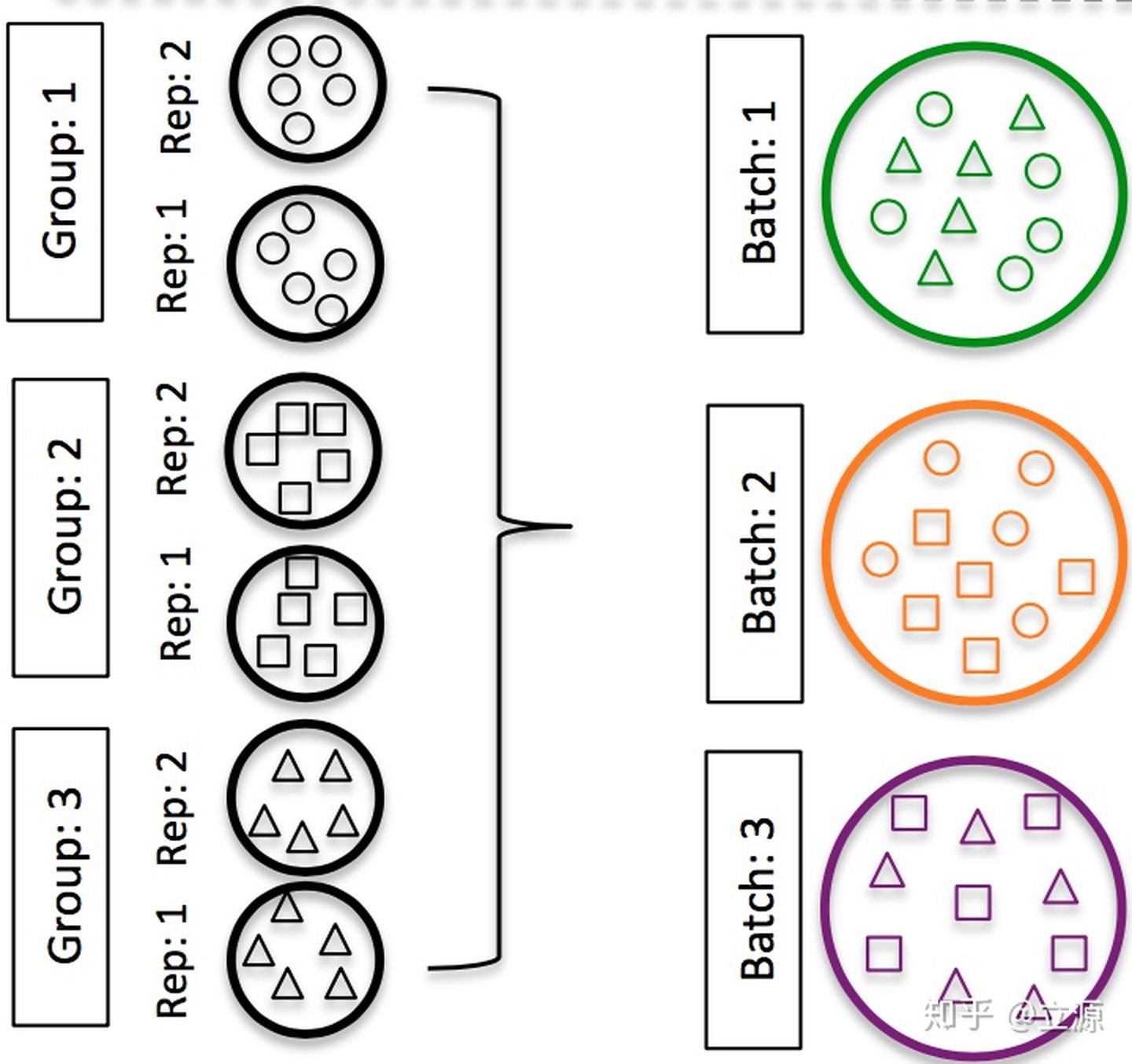
- 実験メタデータにバッチ情報を含めてください。このようにして、分析のバッチによる差異を取り除くことができます。この情報があれば、最終結果に影響はありません。