Seurat 単一細胞トランスクリプトーム シーケンス データ解析チュートリアル (2) - Python (scanpy)
記事内ではscanpy公式サイトを参考に詳しい説明をしています。
このチュートリアルでは、scanpy の視覚化の可能性を探求し、3 つの部分に分かれています。
埋め込み散布図 (UMAP、t-SNE など)
既知のマーカー遺伝子を使用したクラスターの
特定 差次的に発現された遺伝子の視覚化
このチュートリアルでは、PBMC からの 68,000 細胞を含む 10x のデータセットを使用します。Scanpy の配布には、このデータセットの縮小サンプルが含まれています。これには、わずか 700 個の細胞と 765 個の非常に可変性の高い遺伝子が含まれています。データセットは前処理され、UMAP が計算されています。
このチュートリアルでは、次の文献マーカーも使用します。
B 細胞: CD79A、MS4A1
血漿: IGJ (JCHAIN)
T 細胞: CD3D
NK: GNLY、NKG7
骨髄系: CST3、LYZ
単球: FCGR3A
樹状細胞: FCER1A
埋め込み散布図
import scanpy as sc
import pandas as pd
from matplotlib.pyplot import rc_context
sc.set_figure_params(dpi=100, color_map = 'viridis_r')
sc.settings.verbosity = 1
sc.logging.print_header()
pbmc データセットをロードする
pbmc = sc.datasets.pbmc68k_reduced()
# inspect pbmc contents
pbmc
遺伝子発現およびその他の変数の視覚化
散布図の場合、プロットされる値は引数 color として指定されます。これは、.obs 内の任意の遺伝子または任意の列にすることができます。ここで、.obs は、各観察/セルの注釈を含むデータフレームです。詳細については、AnnData を参照してください。
# rc_context is used for the figure size, in this case 4x4
with rc_context({'figure.figsize': (4, 4)}):
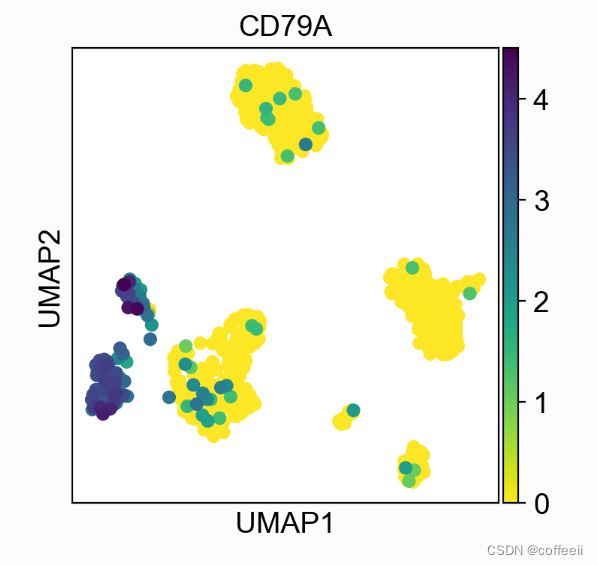
sc.pl.umap(pbmc, color='CD79A')
色の複数の値を指定できます。以下の例では、「CD79A」、「MS4A1」、「IGJ」、「CD3D」、「FCER1A」、「FCGR3A」の 6 つの遺伝子をプロットして、これらのマーカー遺伝子がどこで発現されているかを理解します。
さらに、他の 2 つの値をプロットします。n_counts は各セル (.obs に保存) の UMI カウントであり、bulk_labels は 10X のセルの生のラベルを含むカテゴリ値です。
行ごとのプロットの数は、ncols パラメーターによって制御されます。描画される最大値は、vmax を使用して調整できます (同様に、vmin を最小値に使用できます)。この場合、p99 を使用します。これは、99 パーセンタイルを最大値として使用することを意味します。vmax を複数のパーセルに対して個別に設定したい場合は、最大値を数値または数値のリストにすることができます。
さらに、frameon=False を使用してプロットの周囲のボックスを削除し、s=50 を使用してポイント サイズを設定しています。
with rc_context({'figure.figsize': (3, 3)}):
sc.pl.umap(pbmc, color=['CD79A', 'MS4A1', 'IGJ', 'CD3D', 'FCER1A', 'FCGR3A', 'n_counts', 'bulk_labels'], s=50, frameon=False, ncols=4, vmax='p99')
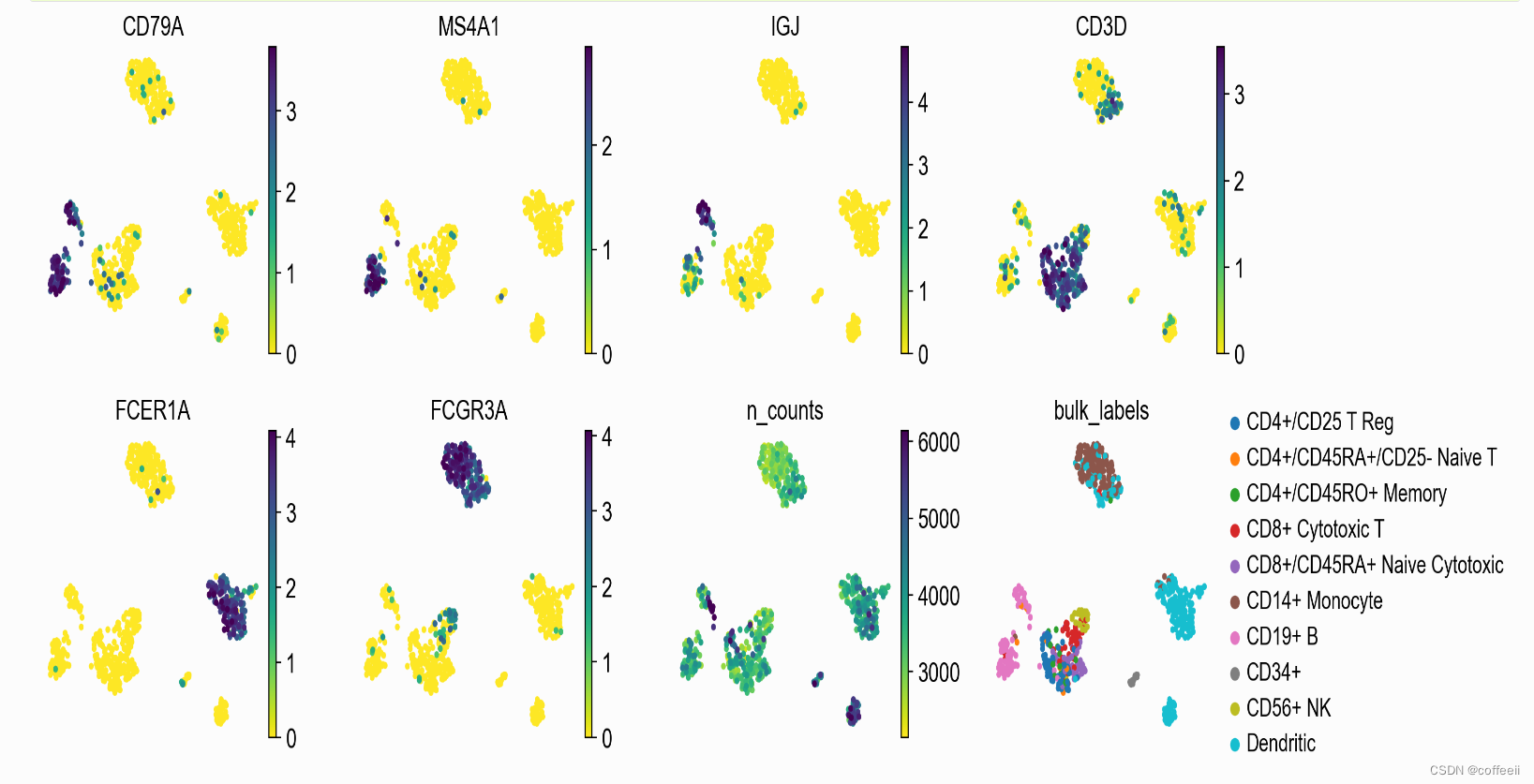
この図では、マーカー遺伝子を発現している細胞のグループと、元の細胞ラベルとの一致がわかります。
散布図関数には、画像を微調整するための多くのオプションがあります。たとえば、次のようにクラスターを表示できます。
# compute clusters using the leiden method and store the results with the name `clusters`
sc.tl.leiden(pbmc, key_added='clusters', resolution=0.5)
with rc_context({'figure.figsize': (5, 5)}):
sc.pl.umap(pbmc, color='clusters', add_outline=True, legend_loc='on data',
legend_fontsize=12, legend_fontoutline=2,frameon=False,
title='clustering of cells', palette='Set1')

既知のマーカー遺伝子に基づくクラスターの同定
通常、クラスターを標識するには、よく知られたマーカー遺伝子が必要です。散布図を使用すると、遺伝子の発現を確認し、それをクラスターに関連付けることができます。ここでは、ドット プロット、バイオリン プロット、ヒート マップ、および「軌跡プロット」と呼ばれるものを使用して、マーカー遺伝子をクラスターに関連付ける追加の視覚的方法を示します。これらの視覚化はすべて同じ情報を要約し、クラスターによって表現されており、最良の結果の選択は研究者に任されています。
まず、ラベル付き遺伝子を含む辞書を構築します。これにより、scanpy がゲノムに自動的にラベルを付けることができるようになります。
marker_genes_dict = {
'B-cell': ['CD79A', 'MS4A1'],
'Dendritic': ['FCER1A', 'CST3'],
'Monocytes': ['FCGR3A'],
'NK': ['GNLY', 'NKG7'],
'Other': ['IGLL1'],
'Plasma': ['IGJ'],
'T-cell': ['CD3D'],
}
ドットプロット
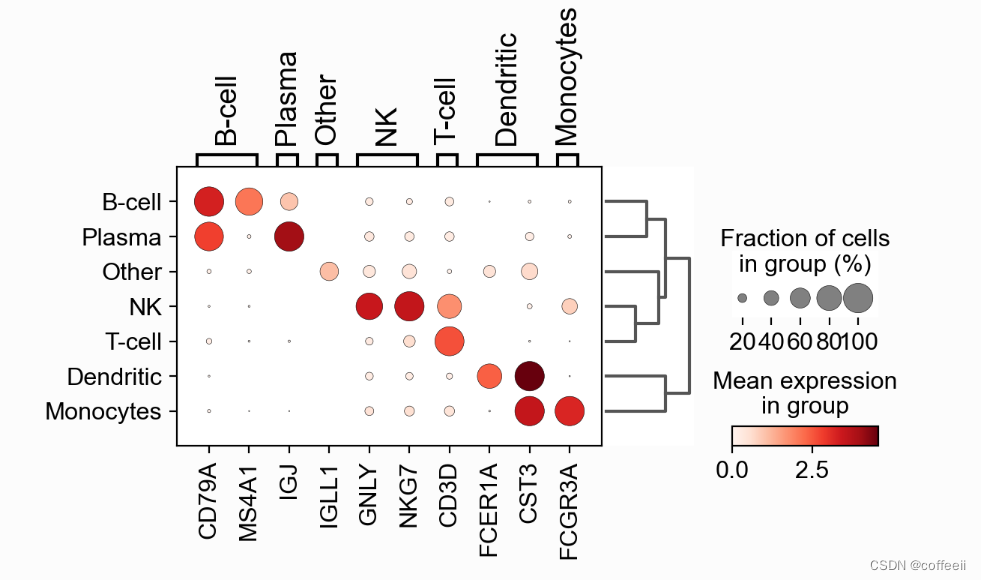
各クラスター内のこれらの遺伝子の発現を調べる簡単な方法は、ドット プロットを使用することです。このタイプのプロットでは、2 種類の情報が要約されます。色は各カテゴリ (この場合は各クラスター) の平均発現を表し、ポイント サイズは遺伝子を発現するカテゴリ内の細胞の割合を表します。
さらに、グラフに樹形図を追加して、類似したクラスターをクラスター化すると便利です。階層的クラスタリングは、クラスタ間の PCA コンポーネントの相関関係を使用して自動的に計算されます。
sc.pl.dotplot(pbmc, marker_genes_dict, 'clusters', dendrogram=True)
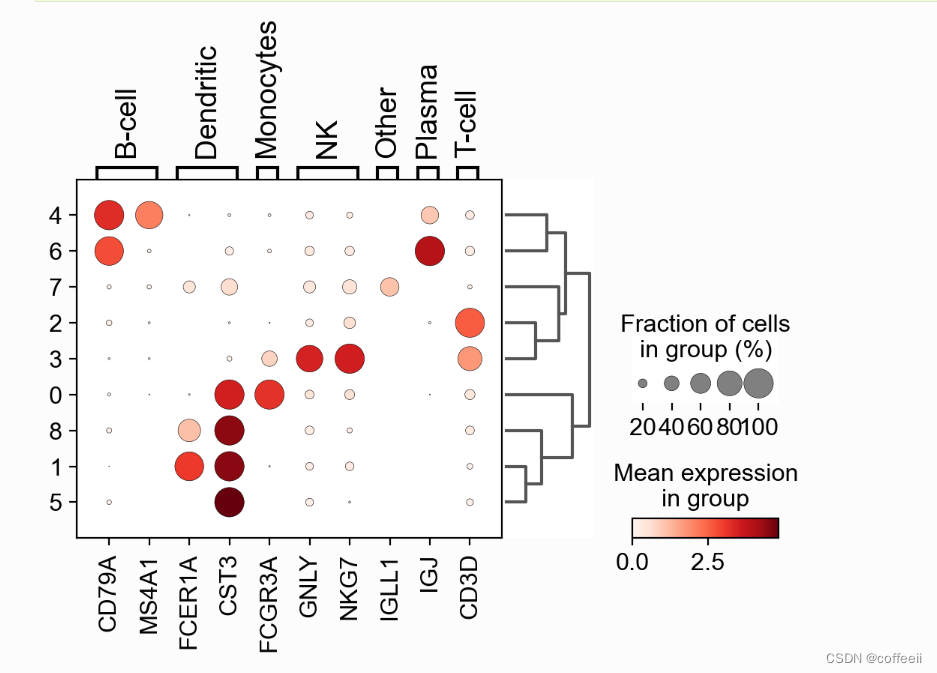
この図を使用すると、クラスター 4 が B 細胞に対応し、クラスター 2 が T 細胞に対応することがわかります。この情報を使用して、次のようにセルに手動で注釈を付けることができます。
# create a dictionary to map cluster to annotation label
cluster2annotation = {
'0': 'Monocytes',
'1': 'Dendritic',
'2': 'T-cell',
'3': 'NK',
'4': 'B-cell',
'5': 'Dendritic',
'6': 'Plasma',
'7': 'Other',
'8': 'Dendritic',
}
# add a new `.obs` column called `cell type` by mapping clusters to annotation using pandas `map` function
pbmc.obs['cell type'] = pbmc.obs['clusters'].map(cluster2annotation).astype('category')
sc.pl.dotplot(pbmc, marker_genes_dict, 'cell type', dendrogram=True)
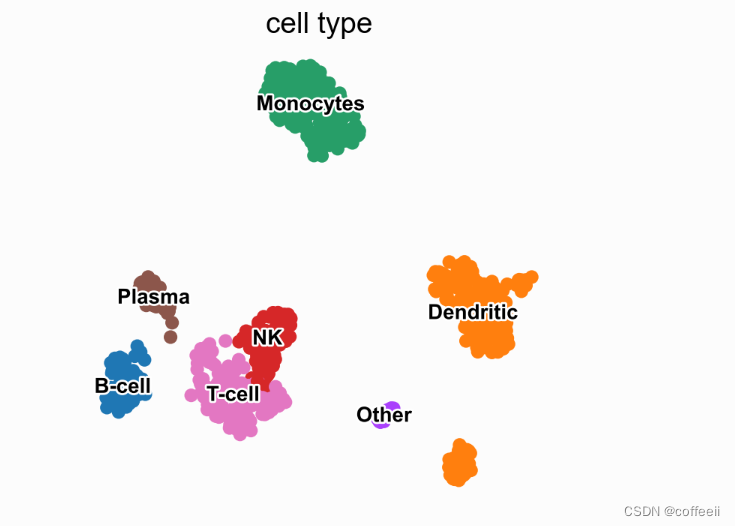
sc.pl.umap(pbmc, color='cell type', legend_loc='on data',
frameon=False, legend_fontsize=10, legend_fontoutline=2)
ヴァイオリンのダイアグラム
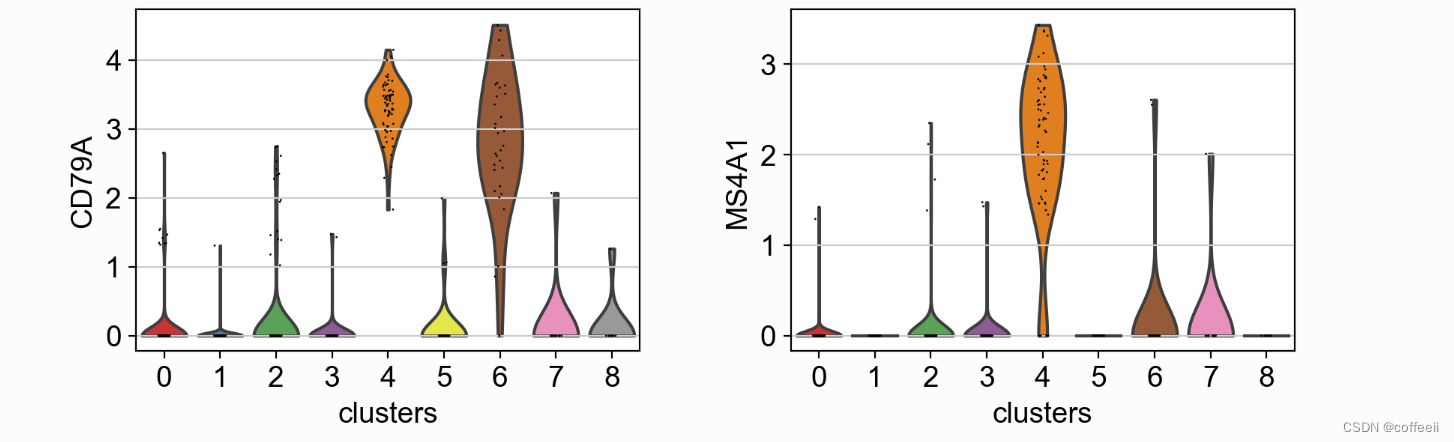
マーカーを探索するもう 1 つの方法は、バイオリン プロットを使用することです。ここでは、クラスター 5 と 8 で CD79A が発現し、クラスター 5 で MS4A1 が発現していることがわかります。ドット プロットと比較して、バイオリン プロットでは、細胞間の遺伝子発現値の分布を把握できます。
with rc_context({'figure.figsize': (4.5, 3)}):
sc.pl.violin(pbmc, ['CD79A', 'MS4A1'], groupby='clusters' )
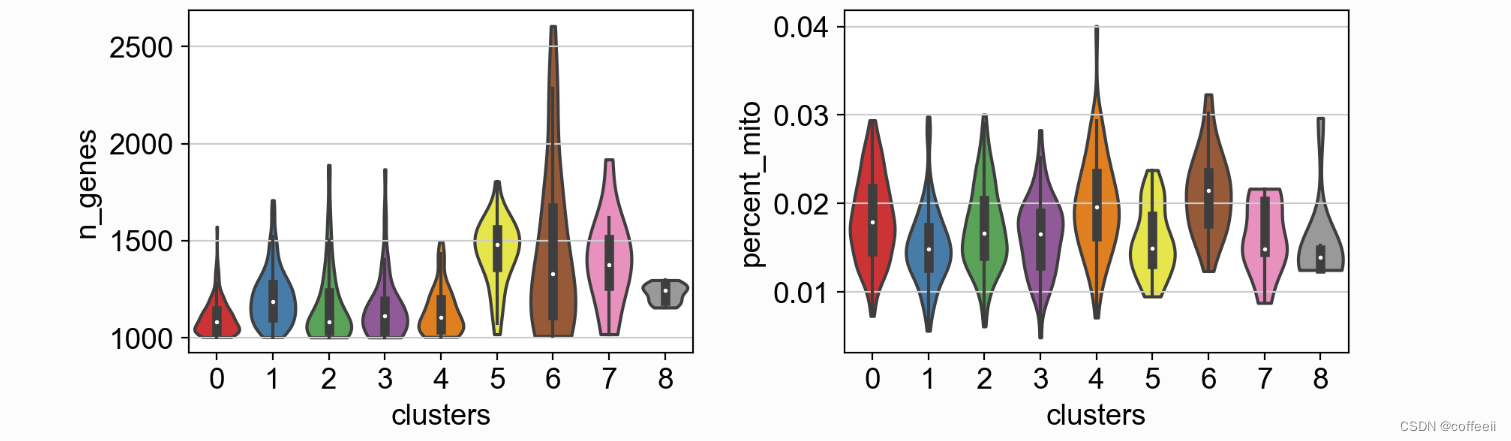
with rc_context({'figure.figsize': (4.5, 3)}):
sc.pl.violin(pbmc, ['n_genes', 'percent_mito'], groupby='clusters', stripplot=False, inner='box') # use stripplot=False to remove the internal dots, inner='box' adds a boxplot inside violins
積み重ねられたバイオリンのプロット
また、使用したすべてのマーカー遺伝子のバイオリン プロット sc.pl.stacked_violin も表示します。以前と同様に、類似したクラスターをグループ化するために樹状図が追加されました。
ax = sc.pl.stacked_violin(pbmc, marker_genes_dict, groupby='clusters', swap_axes=False, dendrogram=True)
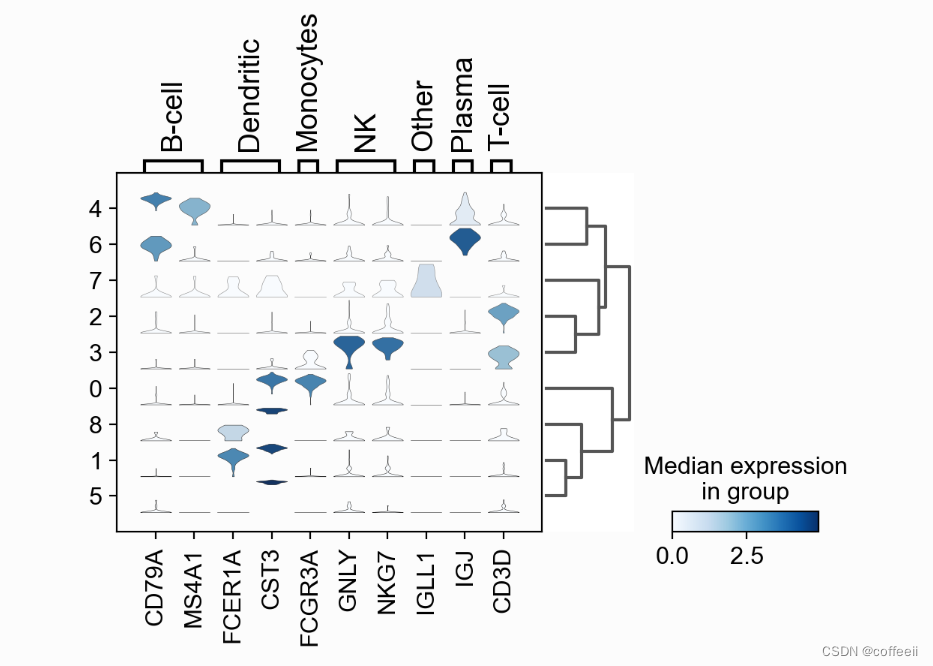
マトリックス図
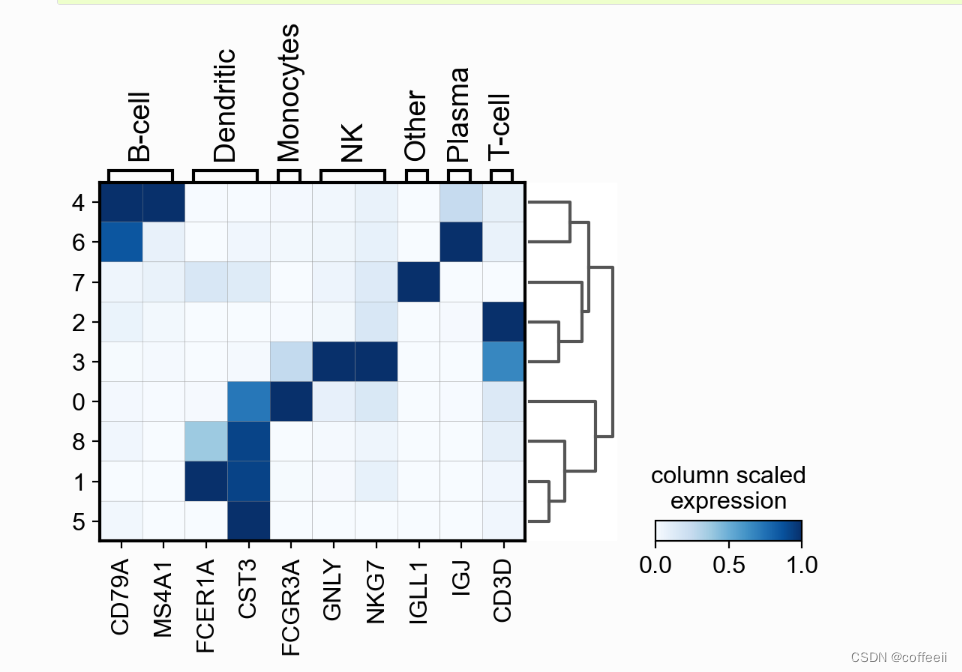
遺伝子発現を視覚化する簡単な方法は、カテゴリ別にグループ化された各遺伝子の平均発現値のヒートマップです。このタイプのプロットでは、基本的にドット プロットの色と同じ情報が表示されます。マトリックスプロット
ここで、遺伝子の発現は 0 から 1 までのスケールで表されます。これは最大の平均発現であり、0 が最小です。
sc.pl.matrixplot(pbmc, marker_genes_dict, 'clusters', dendrogram=True, cmap='Blues', standard_scale='var', colorbar_title='column scaled\nexpression')
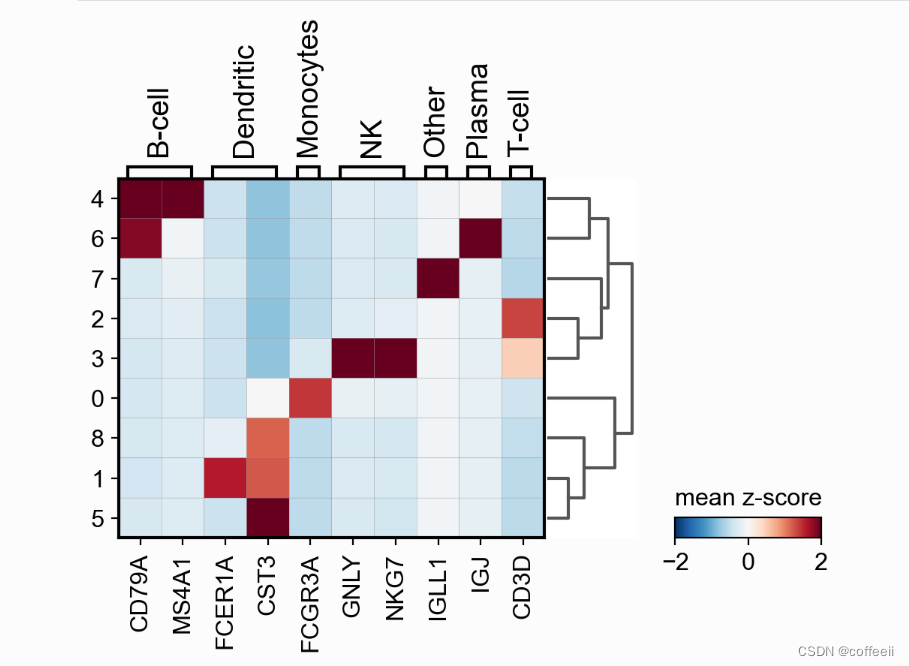
その他の便利なオプションは、sc.pp.scale を使用することです。ここでは、この情報をスケール レイヤーの下に保存します。その後、プロットの最小値と最大値を調整し、発散カラーマップを使用します (この場合、RdBu_r means_r はその逆です)。
# scale and store results in layer
pbmc.layers['scaled'] = sc.pp.scale(pbmc, copy=True).X
sc.pl.matrixplot(pbmc, marker_genes_dict, 'clusters', dendrogram=True,
colorbar_title='mean z-score', layer='scaled', vmin=-2, vmax=2, cmap='RdBu_r')
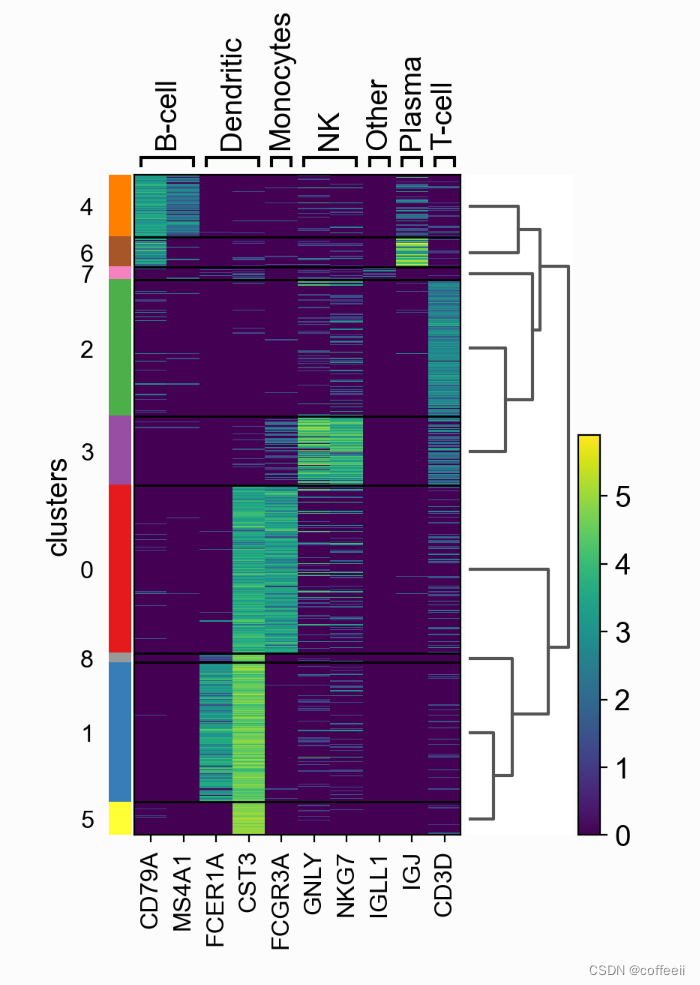
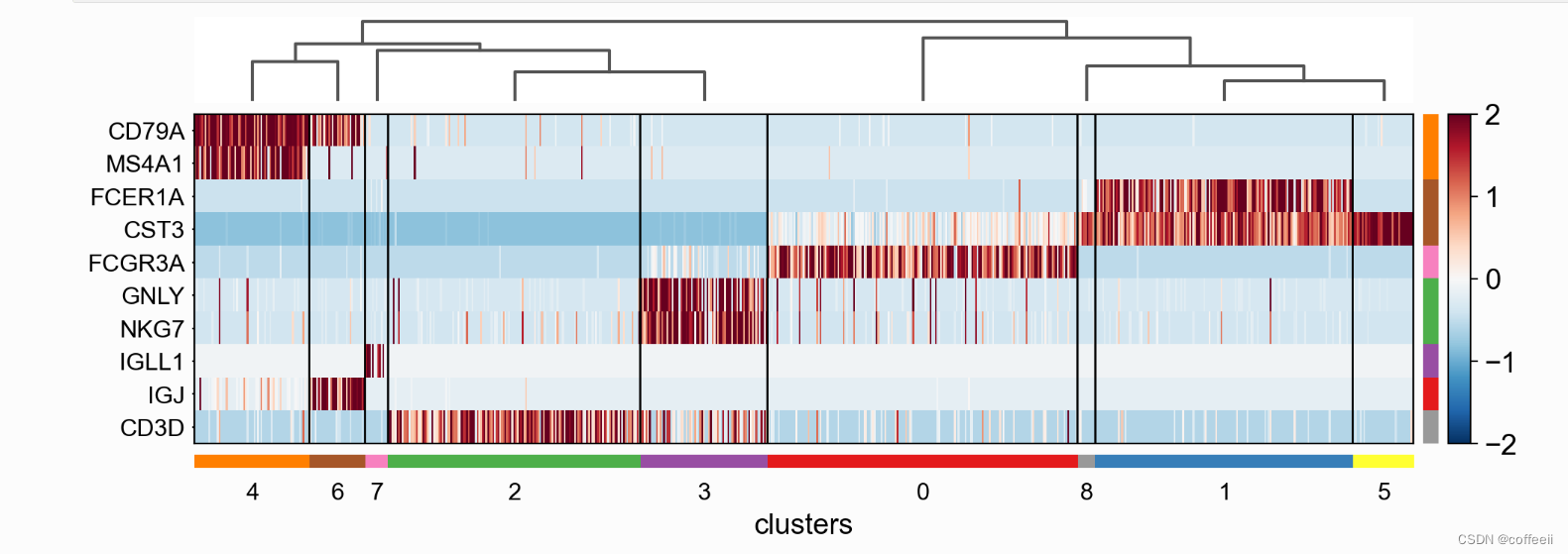
ヒートマップ
ヒート マップでは、前のプロットのようにセルが折りたたまれません。代わりに、各セルが行 (swap_axes=True の場合は列) に表示されます。groupby 情報を追加し、見つかったものと同じカラー コードまたは他の埋め込みを使用して sc.pl.umap を表示できます。
ax = sc.pl.heatmap(pbmc, marker_genes_dict, groupby='clusters', cmap='viridis', dendrogram=True)
ヒートマップは、スケールされたデータ上にプロットすることもできます。次の画像では、前のマトリックス プロットと同様に、最小値と最大値が調整され、発散カラー マップが使用されます。
ax = sc.pl.heatmap(pbmc, marker_genes_dict, groupby='clusters', layer='scaled', vmin=-2, vmax=2, cmap='RdBu_r', dendrogram=True, swap_axes=True, figsize=(11,4))
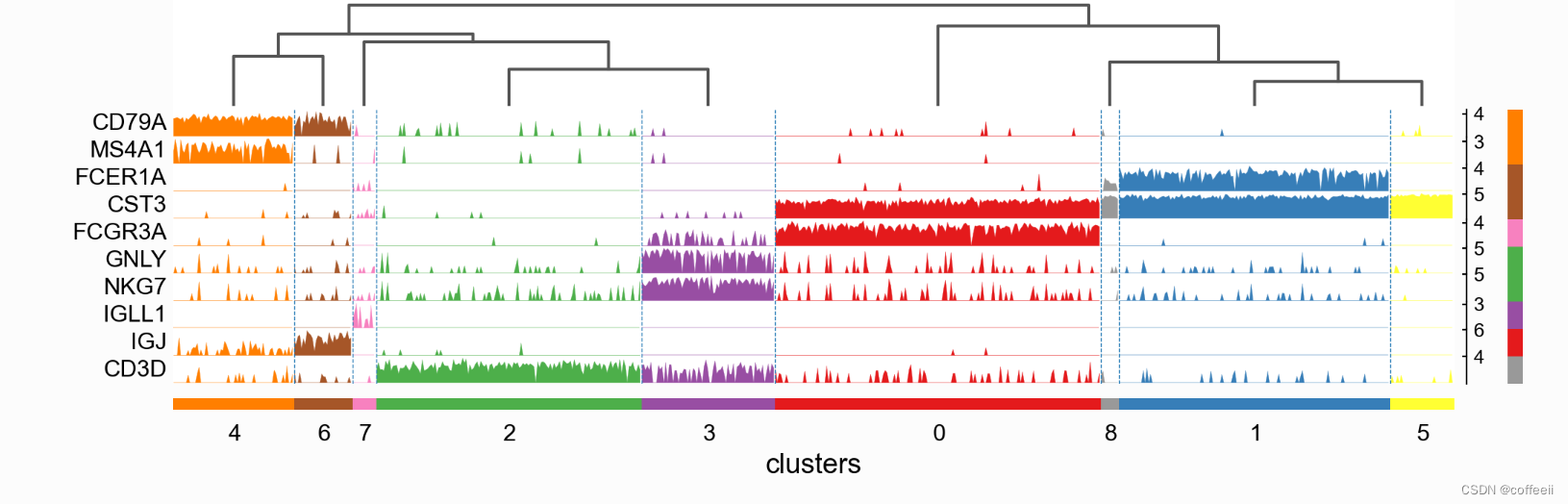
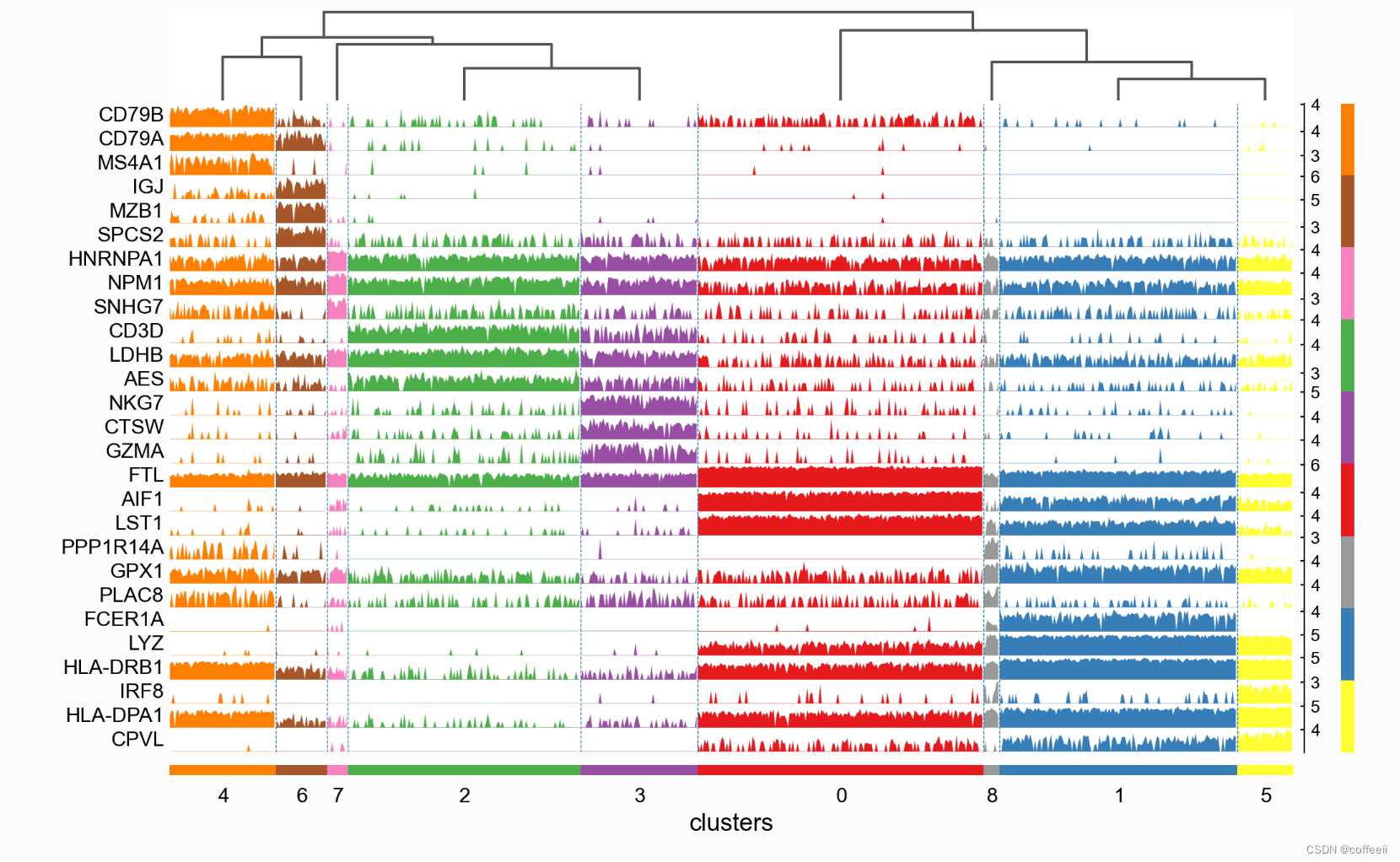
軌跡図
軌跡プロットにはヒート マップと同じ情報が表示されますが、遺伝子発現はカラー スケールではなく高さで表されます。
ax = sc.pl.tracksplot(pbmc, marker_genes_dict, groupby='clusters', dendrogram=True)
マーカー遺伝子の可視化
以前のように既知の遺伝子マーカーによってクラスターを特徴付ける代わりに、クラスターまたはグループ内で差次的に発現される遺伝子を特定できます。
差次的に発現される遺伝子を特定するために、sc.tl.rank_genes_groups を実行しました。この関数は、細胞の各グループを取得し、あるグループ内の各遺伝子の分布を、そのグループに含まれていない他のすべての細胞の分布と比較します。ここでは、10x によって与えられた元の細胞ラベルを使用して、これらの細胞タイプのマーカー遺伝子を特定します。
sc.tl.rank_genes_groups(pbmc, groupby='clusters', method='wilcoxon')
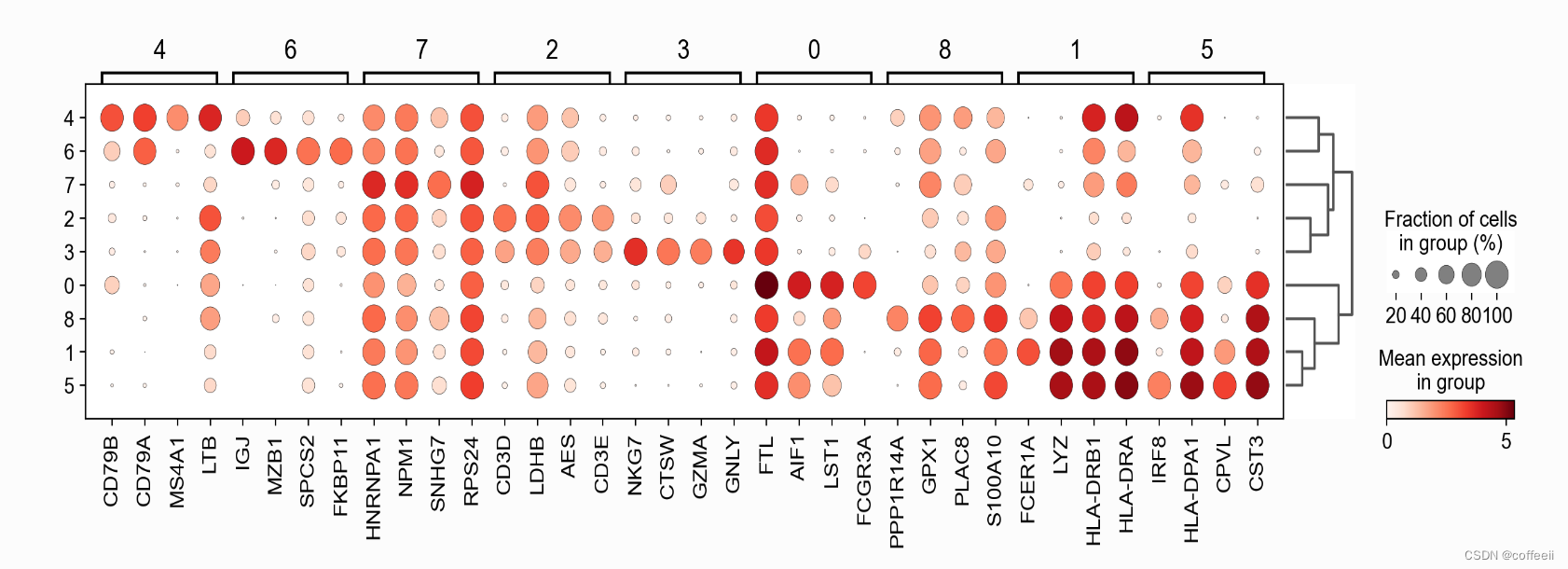
ドットプロットを使用したマーカー遺伝子の視覚化
ドット プロットの視覚化は、差次的な発現を示す遺伝子のプロファイルを理解するのに役立ちます。結果の画像をよりコンパクトにするために、n_genes=4 を使用して上位 4 つのスコア遺伝子のみを表示します。
sc.pl.rank_genes_groups_dotplot(pbmc, n_genes=4)
より適切に表現するには、遺伝子発現の代わりに対数倍率変化をプロットします。さらに、細胞型の発現と残りの細胞の間で対数倍変化が 3 以上である遺伝子に焦点を当てたいと考えました。
この場合、values_to_plot='logfoldchanges' および min_logfoldchange=3 を設定します。
対数倍変化は発散スケールであるため、プロットする最小値と最大値も調整し、発散カラーマップを使用します。下の画像では T 細胞集団を区別するのが難しいことに注意してください。
sc.pl.rank_genes_groups_dotplot(pbmc, n_genes=4, values_to_plot='logfoldchanges', min_logfoldchange=3, vmax=7, vmin=-7, cmap='bwr')
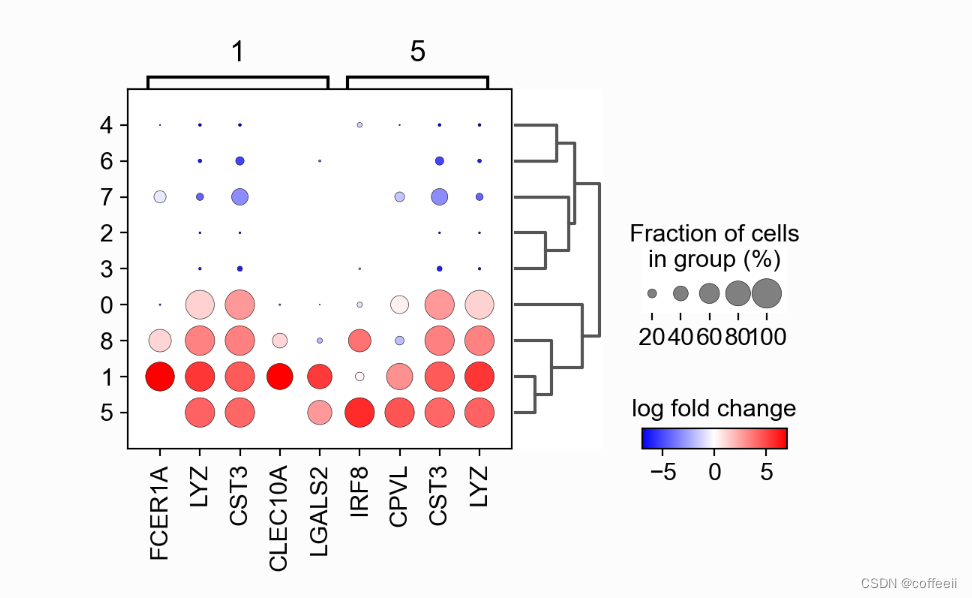
特定のグループに焦点を当てる
次に、2 つのグループのみに焦点を当てたドット プロットを使用します (グループ オプションは、バイオリン プロット、ヒート マップ、およびマトリックス プロットにも使用できます)。この場合、n_genes=30 のすべての遺伝子を最大 30 個表示するように設定します。min_logfoldchange=4
sc.pl.rank_genes_groups_dotplot(pbmc, n_genes=30, values_to_plot='logfoldchanges', min_logfoldchange=4, vmax=7, vmin=-7, cmap='bwr', groups=['1', '5'])
マトリックスプロットを使用してマーカー遺伝子を視覚化する
下の画像では、以前に計算された「スケール」値 (スケールされたレイヤーに保存されている) を使用し、発散カラーマップを使用します。\
sc.pl.rank_genes_groups_matrixplot(pbmc, n_genes=3, use_raw=False, vmin=-3, vmax=3, cmap='bwr', layer='scaled')
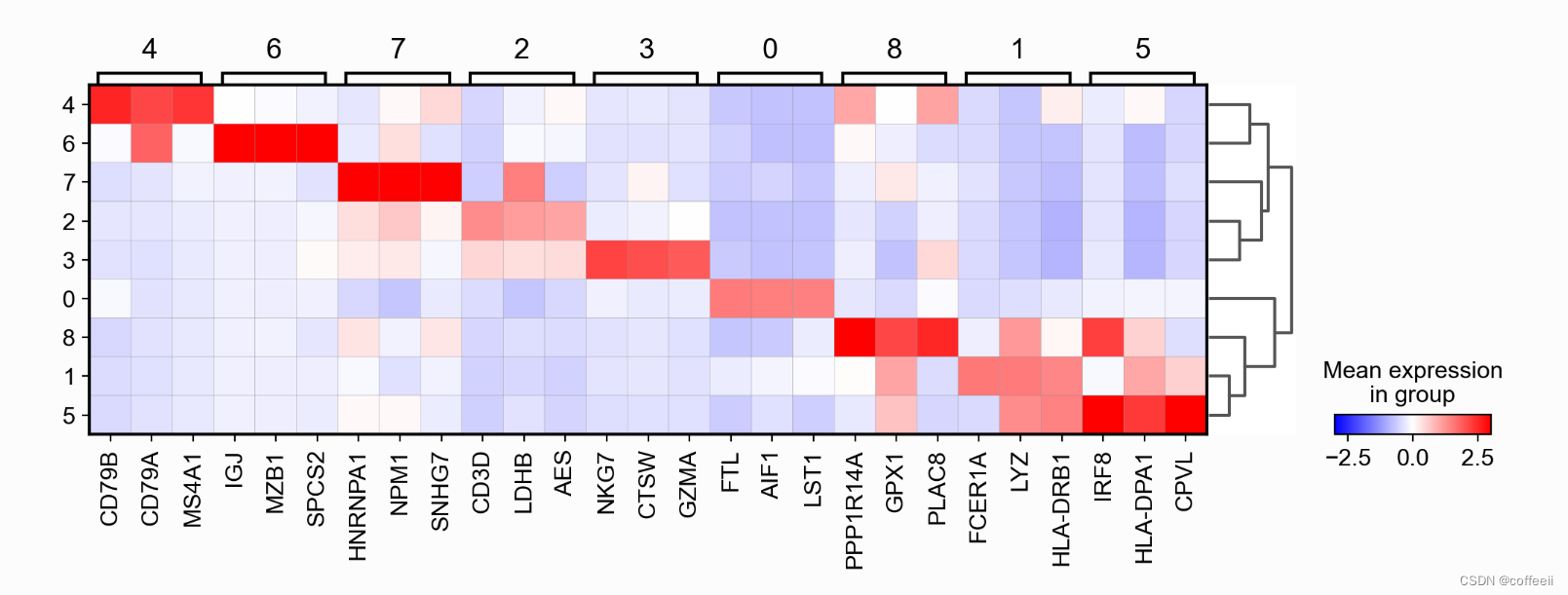
積み上げバイオリンプロットを使用したマーカー遺伝子の視覚化
sc.pl.rank_genes_groups_stacked_violin(pbmc, n_genes=3, cmap='viridis_r')
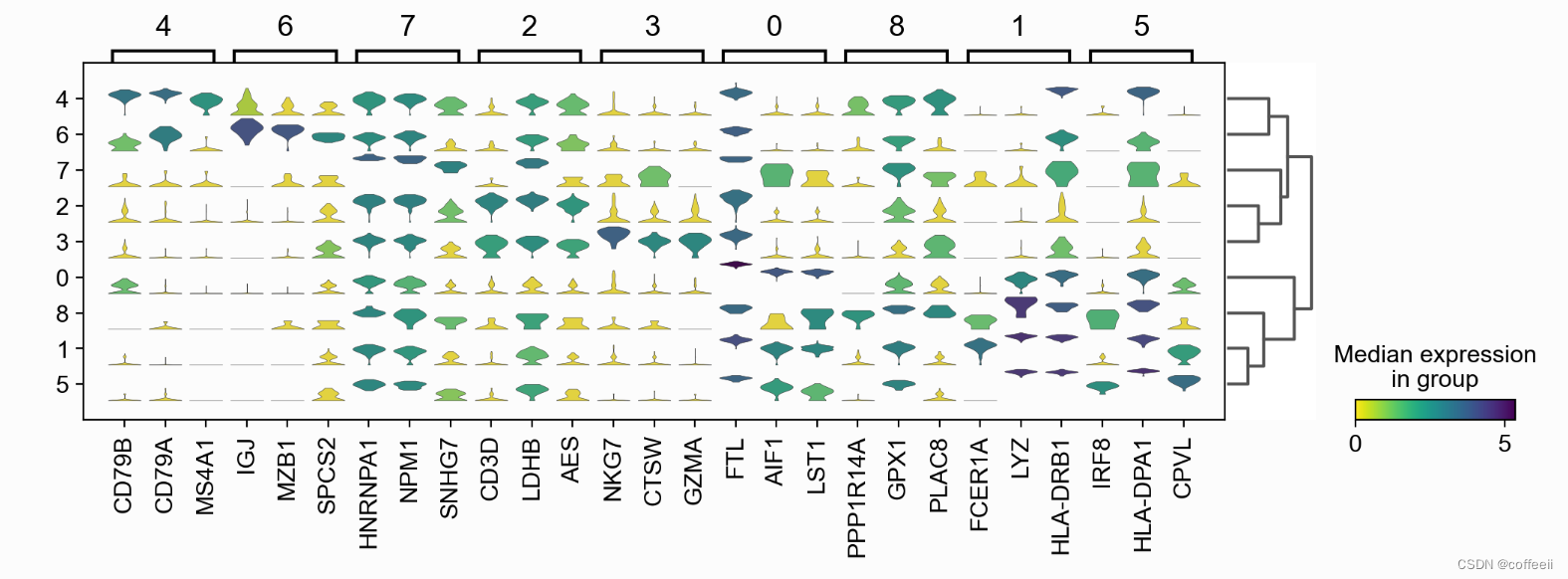
ヒートマップを使用したマーカー遺伝子の視覚化
sc.pl.rank_genes_groups_heatmap(pbmc, n_genes=3, use_raw=False, swap_axes=True, vmin=-3, vmax=3, cmap='bwr', layer='scaled', figsize=(10,7), show=False);
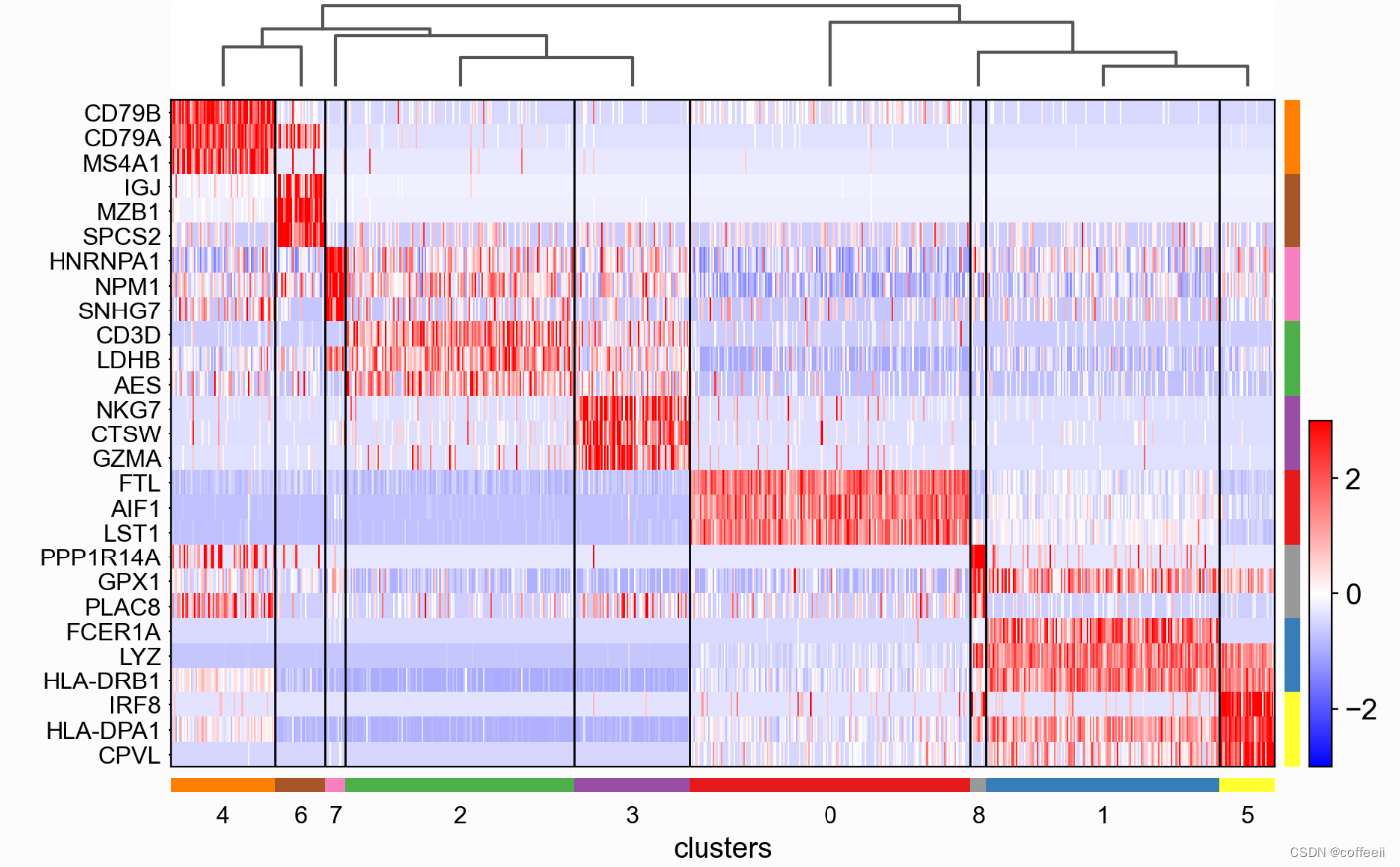
カテゴリごとに 10 個の遺伝子が表示されます。遺伝子ラベルはオフになり、軸が交換されます。画像を交換すると、「括弧」の代わりにカテゴリのカラーコードが表示されることに注意してください。
sc.pl.rank_genes_groups_heatmap(pbmc, n_genes=10, use_raw=False, swap_axes=True, show_gene_labels=False,
vmin=-3, vmax=3, cmap='bwr')
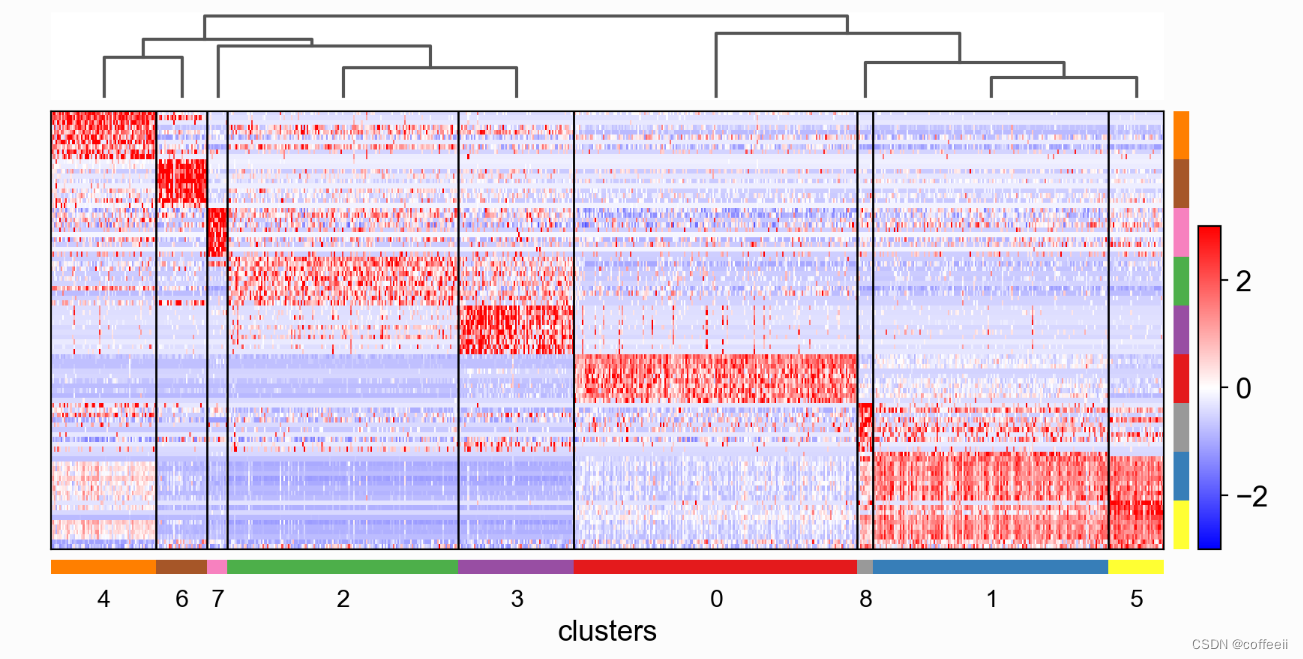
軌跡プロットを使用したマーカー遺伝子の視覚化
sc.pl.rank_genes_groups_tracksplot(pbmc, n_genes=3)
ツリーマップのオプション
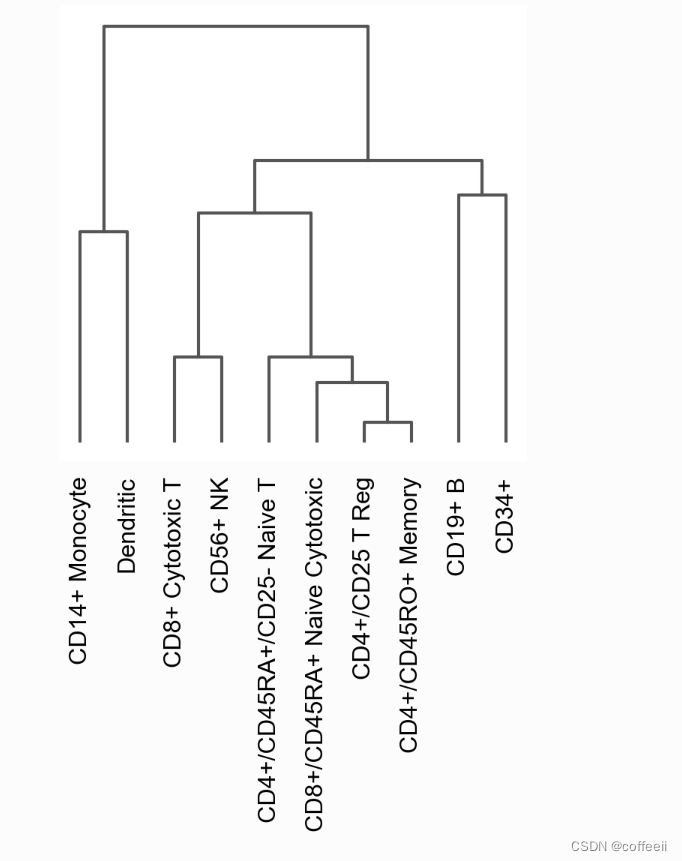
ほとんどのビジュアライゼーションでは、ツリー図を使用してカテゴリを整理できます。ただし、次のように樹状図を独立して描画することもできます。
# compute hierarchical clustering using PCs (several distance metrics and linkage methods are available).
sc.tl.dendrogram(pbmc, 'bulk_labels')
ax = sc.pl.dendrogram(pbmc, 'bulk_labels')