Este número presenta principalmente algunos conceptos básicos del análisis de enriquecimiento funcional de genes y también presenta cómo utilizar la herramienta de análisis en línea DAVID para realizar análisis de enriquecimiento funcional de genes GO / KEGG.
¿Qué es el análisis de enriquecimiento funcional genético?
El análisis de enriquecimiento de la función genética se refiere al análisis estadístico y la clasificación de la función genética de muchos genes obtenidos a través de varias bases de datos, extrayendo así las categorías de funciones genéticas en la base de datos que están significativamente relacionadas con los problemas biológicos que estamos estudiando. Sin embargo, cabe señalar que diferentes combinaciones de genes pueden tener diferentes funciones biológicas en diferentes entornos biológicos. Por lo tanto, es necesario seleccionar los conjuntos de genes correspondientes de acuerdo con la situación real y conectarlos con los cambios funcionales relacionados con el objeto de estudio, para clasificar los muchos genes diferenciales y finalmente centrarse en los genes diferenciales clave relacionados con la biología. problema en estudio, proporcionando así dirección y base para la verificación experimental posterior. Con todo, la esencia del análisis de enriquecimiento de la función genética es el análisis de conglomerados, que se utiliza para interpretar el conocimiento biológico detrás de un grupo de genes y revelar sus funciones dentro o fuera de la célula.
¿Por qué realizar análisis de enriquecimiento funcional?
Con el desarrollo de la tecnología de secuenciación de alto rendimiento, el campo de la investigación biológica ha entrado en la era de las ómicas. Sin embargo, la enorme cantidad de datos en secuenciación ómica ha intimidado a los investigadores. La extracción y el análisis eficaces de esta información también se ha convertido en un foco clave de muchos investigadores. Tomando como ejemplo la secuenciación del transcriptoma (RNA-seq), los resultados de la secuenciación a menudo producen una serie de genes expresados diferencialmente, pero la forma en que los investigadores conectan estos genes con las cuestiones biológicas que se estudian y los posibles mecanismos reguladores se ha convertido en una cuestión clave. Por lo tanto, los investigadores pueden realizar análisis de enriquecimiento funcional en genes a través de múltiples bases de datos de anotaciones funcionales, dividir esta serie de conjuntos de genes en diferentes categorías funcionales y buscar vías biológicas que desempeñen un papel clave en los procesos biológicos, revelando y comprendiendo así las moléculas básicas y subyacentes. Mecanismos de estos procesos biológicos. De hecho, el punto de partida del nivel molecular es el nivel genético, pero hay muchos tipos de genes y la mejor manera de comprender el significado biológico de estos genes es el análisis de enriquecimiento de la función genética .
Dependiendo de la base de datos de selección y anotación de genes durante el proceso de análisis de enriquecimiento, los análisis de enriquecimiento comúnmente utilizados se pueden dividir en los siguientes tipos: enriquecimiento funcional GO, enriquecimiento de la vía KEGG, enriquecimiento del conjunto de genes GSEA, etc.
Análisis de enriquecimiento funcional GO.
La base de datos Gene Ontology (GO) es una base de datos creada por el Consorcio GO en 2000. Su objetivo es establecer una base de datos que sea adecuada para diversas especies, defina y describa las funciones de genes y proteínas, y pueda continuar evolucionando con la investigación. -Profundidad y estándares de vocabulario semántico actualizados. La anotación GO cubre tres aspectos: función biológica molecular ( MF ) , proceso biológico ( BP) y componentes celulares ( CC ). A través funcionestresde estas La anotación GO es uno de los sistemas de anotación de genes más utilizados actualmente. La base de conocimientos de GO es la fuente de información más grande del mundo sobre la función de los genes. Este conocimiento es legible tanto por humanos como por máquinas, y es la base para el análisis computacional de experimentos de biología molecular y genética a gran escala en la investigación biomédica.
Función molecular (MF):
Describe la función o funciones de los genes a nivel de biología molecular, como actividad catalítica, actividad de transporte, actividad de unión, etc. La función molecular se refiere principalmente a la función de un solo producto genético, y una pequeña parte se refiere a la función del complejo formado por este producto genético.
Proceso biológico (BP):
Describe los procesos biológicos en los que participa el gen, como la participación en la regulación transcripcional, el procesamiento de ARNr, la replicación del ADN, el crecimiento y mantenimiento celular, la transducción de señales y el transporte de diversos factores. Un proceso biológico es un proceso que consta de funciones moleculares de manera ordenada y tiene múltiples pasos. Una vía biológica no es exactamente lo mismo que una vía biológica. Por lo tanto, GO no implica el complejo proceso de regulación del mecanismo en la vía.
Componente celular (CC):
Describir la ubicación de un gen (producto) en una célula, ya sea en el citoplasma, núcleo, orgánulos, membrana mitocondrial o matriz. O en algunos productos genéticos, como el proteosoma, etc.
RECURSO DE ONTOLOGÍA GENÉTICA: http://geneontology.org/
Enriquecimiento de la vía KEGG
La Enciclopedia de genes y genomas de Kyoto (KEGG) es una base de datos para el análisis sistemático de la función genética y la información del genoma. Integra información de datos de genómica, bioquímica y ómica funcional de sistemas, incluidas las vías metabólicas (KEGG). PATHWAY), medicamentos ( KEGG DRUG ) , enfermedades (ENFERMEDAD DE KEGG), modelos funcionales (MÓDULO KEGG), secuencias genéticas (GENES KEGG) y genomas (GENOMA KEGG), etc. El sistema KO (KEGG ORTHOLOG) vincula varios sistemas de anotaciones KEGG. KEGG ha establecido un sistema completo de anotación KO que puede completar la anotación funcional del genoma o transcriptoma de especies recién secuenciadas. KEGG ayuda a los investigadores a estudiar genes y la información de expresión en su conjunto.
KEGG: https://www.kegg.jp/
Análisis de enriquecimiento GSEA :
El análisis de enriquecimiento de conjuntos de genes (GSEA) generalmente analiza si un grupo de genes está sobrepresentado en un determinado nodo funcional en comparación con niveles aleatorios . El análisis GSEA incluye todos los genes y puede tener en cuenta algunos efectos débiles pero no significativos . El análisis GSEA no requiere análisis diferencial y puede utilizar directamente información de expresión para encontrar vías/conjuntos de genes funcionales relacionados con rasgos . De esta manera, se puede retener cierta información clave sin filtrar, y luego genes funcionales sin diferencias obvias pero con diferencias genéticas consistentes. Se pueden encontrar tendencias.
En los próximos números, presentaré principalmente el uso de herramientas de análisis en línea DAVID , paquetes R clusterProfiler , etc. para realizar análisis de enriquecimiento funcional GO y KEGG y la visualización correspondiente de genes.
La herramienta de análisis en línea DAVID realiza análisis de enriquecimiento funcional de genes GO/KEGG
Paso 1-2
Primero abra el sitio web oficial de DAVID: DAVID Functional Annotation Bioinformatics Microarray Analysis y haga clic en "Anotación de función".
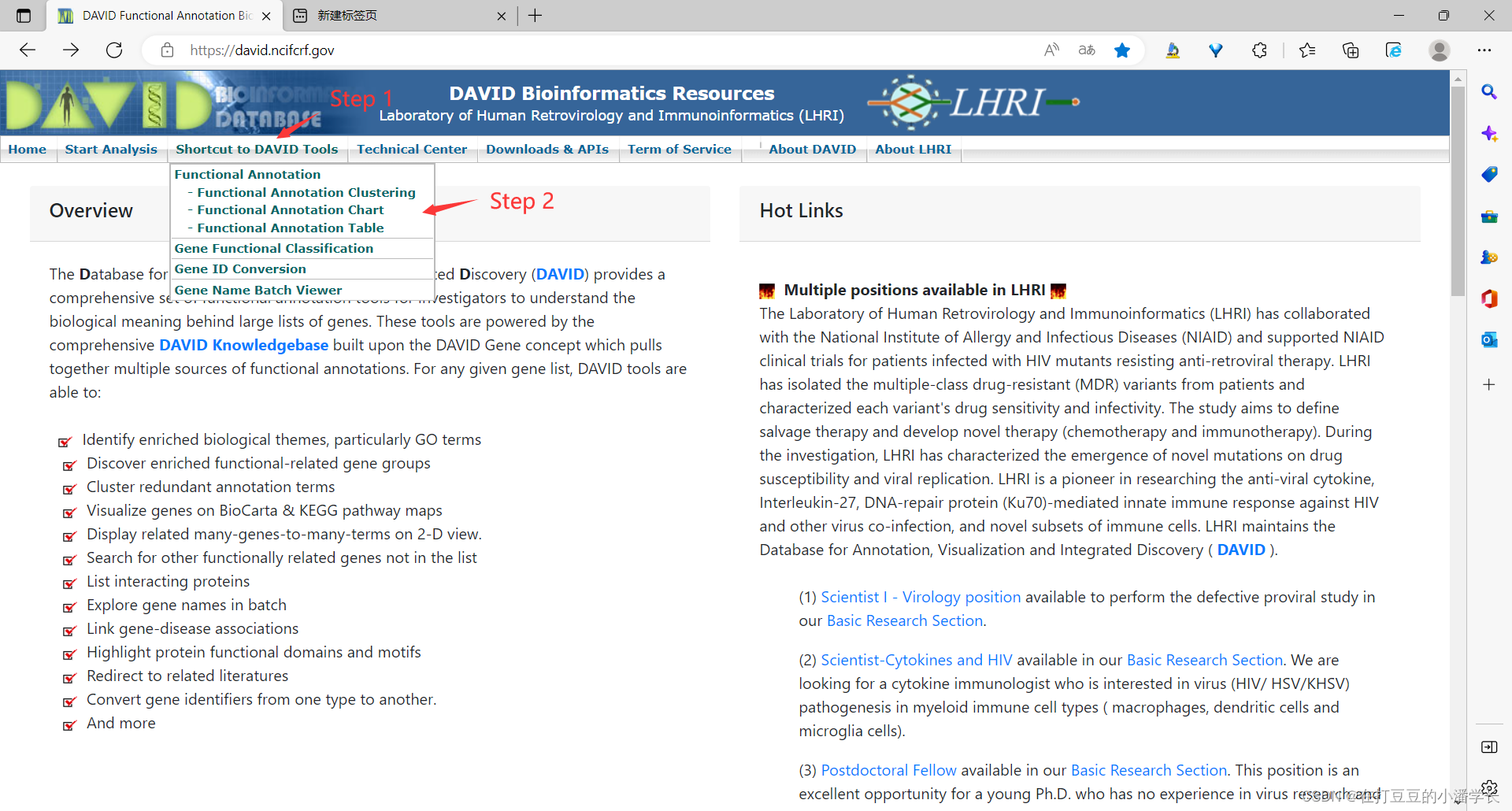
Paso 3
Importar datos: (1) Pegar directamente en "Pegar una lista"; (2) Importar archivos directamente en "Elegir de un archivo", compatible con el formato txt.
 Etapa 4
Etapa 4
Seleccione su tipo de gen en "Seleccionar identificador". Cargué el nombre del gen (símbolo del gen), así que seleccioné "OFFICIAL_GENE_SYMBOL". (Este paso depende principalmente del tipo de datos que importe)
Paso 5
Selecciona la especie que estás estudiando en "Seleccionar especie", yo soy el que estoy estudiando aquí, así que elegí "Homo sapiens".
Paso 6
Seleccione el tipo de lista que se ingresará en "Tipo de lista". Lo que ingresé aquí es el gen en estudio, por lo que seleccioné "Lista de genes".
Paso 7
Haga clic en "Enviar lista" para ejecutar

Paso 8
Ver los resultados del análisis de enriquecimiento de datos
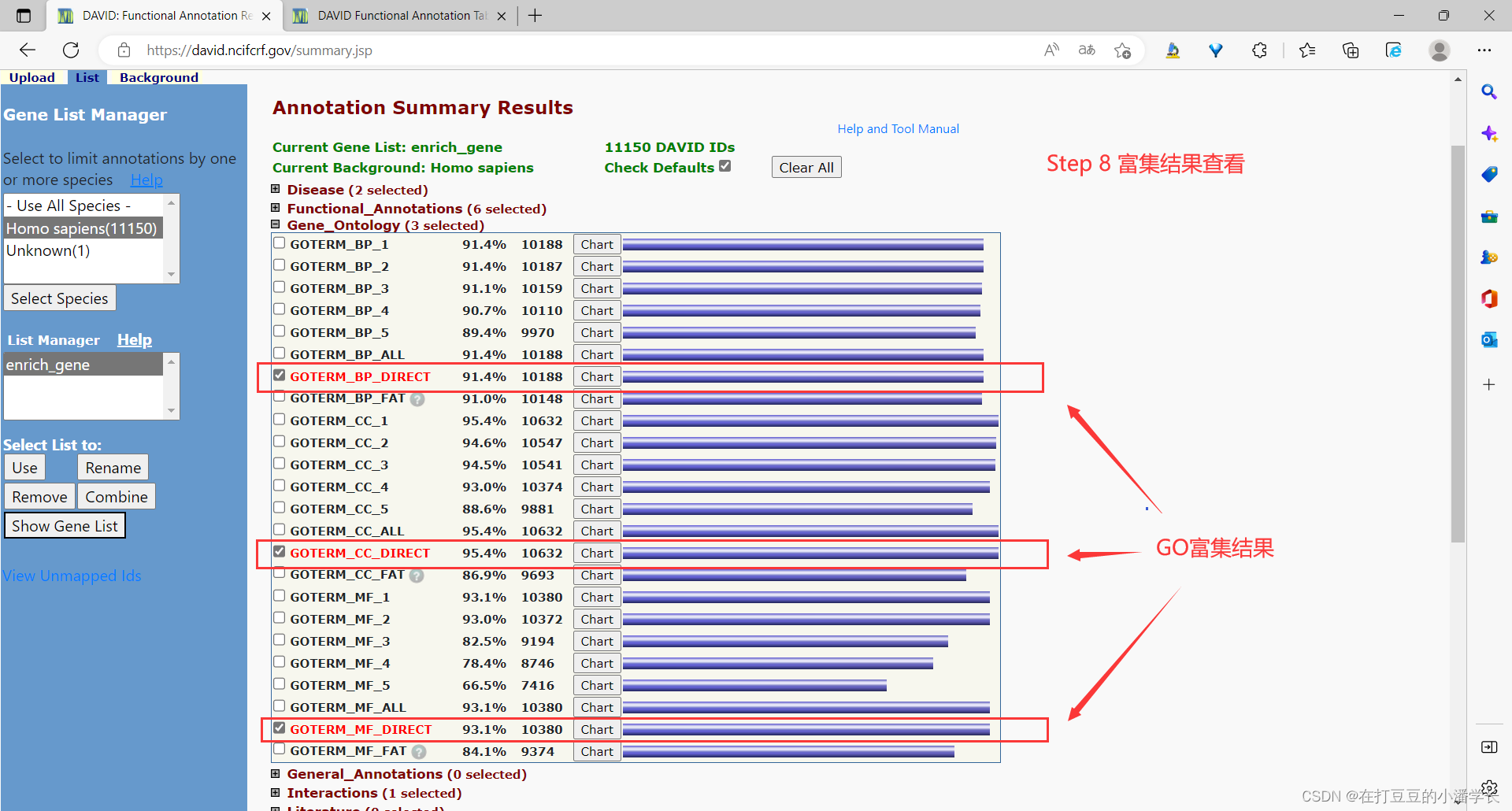
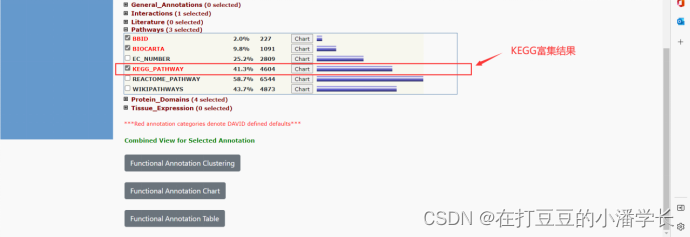
Paso 9
Exportar resultados del análisis de enriquecimiento (copiar y pegar en Excel)

Los resultados de enriquecimiento obtenidos por DAVID consisten principalmente en estas columnas de datos: Categoría, Término (semántica GO), Recuento (número de genes), % (proporción de genes), Valor P (valor P), Genes (nombre del gen), Lista Total, Pop Hits, Pop Total, Fold Enrichment, Bonferroni ( corrección de pruebas múltiples ), Benjamini ( corrección de pruebas múltiples ) y FDR (valor P corregido).
 referencias
referencias
[1] Sherman, BT y cols. DAVID: un servidor web para análisis de enriquecimiento funcional y anotación funcional de listas de genes (actualización de 2021). Ácidos nucleicos res. 50, W216-221, doi:10.1093/nar/gkac194 (2022).
Bueno, este intercambio termina aquí. En el próximo número, compartiremos el método para visualizar estos resultados de enriquecimiento funcional, así que estad atentos.

Siga la cuenta pública de "Senior Xiao Pan Playing Doudou" para obtener más información útil.