Biological network analysis with deep learning(使用深度学习的生物网络分析)很少有关于生物网络的综述类论文,这是今年发在 Briefings in Bioinformatics期刊上的一篇。随着深度学习的发展及广泛应用,这篇论文可以读读看。
摘要
实验高通量技术的最新进展扩大了生物学中分子数据的可用性和数量。鉴于生物过程中相互作用的重要性,例如蛋白质之间的相互作用或化合物中的键,这些数据通常以生物网络的形式表示。这种数据的增长产生了对分析网络的新计算工具的需求。该领域的一个主要趋势是使用深度学习来实现这一目标,更具体地说,使用与网络一起工作的方法,即所谓的图形神经网络(GNNs)。在这篇文章中,我们描述了生物网络,并回顾了神经网络的原理和基本算法。然后我们讨论生物信息学中目前图形神经网络经常应用的领域,如蛋白质功能预测、蛋白质相互作用预测和电子药物发现和开发。最后,我们强调了基因调控网络和疾病诊断等应用领域,在这些领域中,深度学习作为一种新工具正在出现,以回答经典问题,如基因相互作用预测和从数据中自动预测疾病。
一、Introduction
理解许多生物过程不仅需要了解生物实体本身,还需要了解它们之间的关系。例如,细胞分化等过程不仅取决于存在哪些蛋白质,还取决于哪些蛋白质结合在一起。表示这种过程的一种自然方式是作为一个图,也称为网络,因为一个图可以建模两个实体以及它们的交互。
实验高通量技术的最新进展以较低的成本极大地增加了交互屏幕的数据输出,并产生了大量此类生物网络数据[1]。这些数据的可用性使得使用生物网络分析来解决生物信息学中的许多令人兴奋的挑战成为可能,比如预测新蛋白质的功能基于其结构或预测新药将如何与生物途径相互作用。这些丰富的新数据,加上计算技术的最新进展,使得这些数据的快速处理成为可能[2,第440页],重新点燃了人们对神经网络[3–6]的兴趣,这可以追溯到20世纪70年代和80年代,并为深度神经网络的出现奠定了基础,也就是深度学习,作为解决这些未解决问题的新方法。
深度学习是一个由多层(通常是非线性的)激活函数组成的神经网络,其组成能够模拟非线性依赖关系。这已经在多个领域显示出经验上的强大性能,例如图像分析[7]和语音识别[8]。深度学习的优势之一是它能够检测数据中的复杂模式,这使得它非常适合于生物信息学的应用,在生物信息学中,数据代表了生物实体和过程之间复杂的、相互依赖的关系,这些实体和过程通常具有内在的噪声,并且发生在多个尺度上[9]。此外,深度学习方法已经扩展到图结构数据,使其成为解决这些生物网络分析问题的有前途的技术。本文详述的将深度学习应用于生物网络数据的早期例子一直报告了比现有经典机器学习方法可比拟或更好的结果,突出了其在该领域的潜力。
我们从介绍生物网络和描述网络上的典型学习任务开始这篇论文。随后,我们将解释支撑图的深度学习的核心概念,即图神经网络。最后,我们将讨论GNNs在生物信息学中最受欢迎的应用任务。
1. Biological networks
脱氧核糖核酸、核糖核酸、蛋白质和代谢物在生命细胞过程的分子机制中起着至关重要的作用。研究它们的结构和相互作用是很重要的,原因有很多,包括新药的开发和疾病途径的发现。这些实体的结构和相互作用都可以用一个图来表示,该图由一组节点和一组表示节点之间连接的边组成。例如,分子可以表示为一个图形,其中节点是原子,边是原子之间的键。类似地,许多生物过程可以用作为节点的实体和作为边的它们之间的相互作用或关系来建模。由于各种原因,上述图形表示是方便的。网络为异构和复杂的生物过程提供了简单直观的表示[10]。此外,它通过使用图论、机器学习和深度学习技术来促进建模和理解复杂的分子机制。
如上所述,可以在不同的细节层次上定义生物网络。除了用于研究分子特性和功能的生物因素的图形表示之外,其他常见的生物网络包括蛋白质-蛋白质相互作用(PPI)网络、基因调节网络(GRN)和代谢网络。此外,由于它们在当代健康研究中的相关性,上述生物网络的定义被扩展到包括药物-药物相互作用(DDI)网络。在下文中,我们将简要介绍这些网络。
Protein-Protein Interaction Networks PPI网络代表蛋白质之间的相互作用[11]。PPIs对几乎所有细胞功能都是必不可少的[12],从细胞结构成分的组装,即细胞骨架,到转录、翻译和主动转运等过程[13]。蛋白质相互作用还包括短暂的相互作用,即容易形成和破坏的蛋白质复合物[14]。在PPI网络中,节点对应于蛋白质,而边缘定义了连接蛋白质之间的相互作用[15]。PPI的详尽图表示还包括相互作用的类型,即磷酸化或键。然而,在实践中,这很少被捕捉到。
Gene Regulatory Networks GRN代表了调节基因表达的复杂机制,这是一组导致从DNA序列产生蛋白质的过程[16]。调节机制发生在DNA产生蛋白质的不同阶段,如转录、翻译和剪接阶段。对这些复杂且相互关联的机制的直观解释是,蛋白质既是基因表达的产物,也是基因表达的控制者[13]。在GRNs中,每个节点代表一个基因,两个基因之间的直接联系意味着一个基因直接调节另一个基因的表达,而没有其他基因的介导[17]。
Metabolic Networks 代谢网络使用图表来表示新陈代谢,新陈代谢是生物体内发生的维持生命的所有化学反应的集合。代谢作用者称为代谢物,代表代谢反应的中间产物和最终产物。鉴于其复杂性,代谢网络通常被分解为代谢途径,即与执行特定代谢功能相关的一系列化学反应[18]。代谢的图形表示包括将每种代谢物映射到一个节点,将每种反应映射到以酶作为催化剂的有向边[19]。
Drug–Drug Interaction Networks DDI网络的目标是模拟不同药物之间的相互作用[20]。DDI网络以节点的形式提供药物,以边的形式表示药物之间的相互作用。与以前的网络不同,DDI网络不代表生物过程。然而,由于它是药物相互作用知识的有意义的表示,DDI网络现在对研究人员越来越感兴趣。事实上,DDI网络被广泛用于多药物研究[21]。
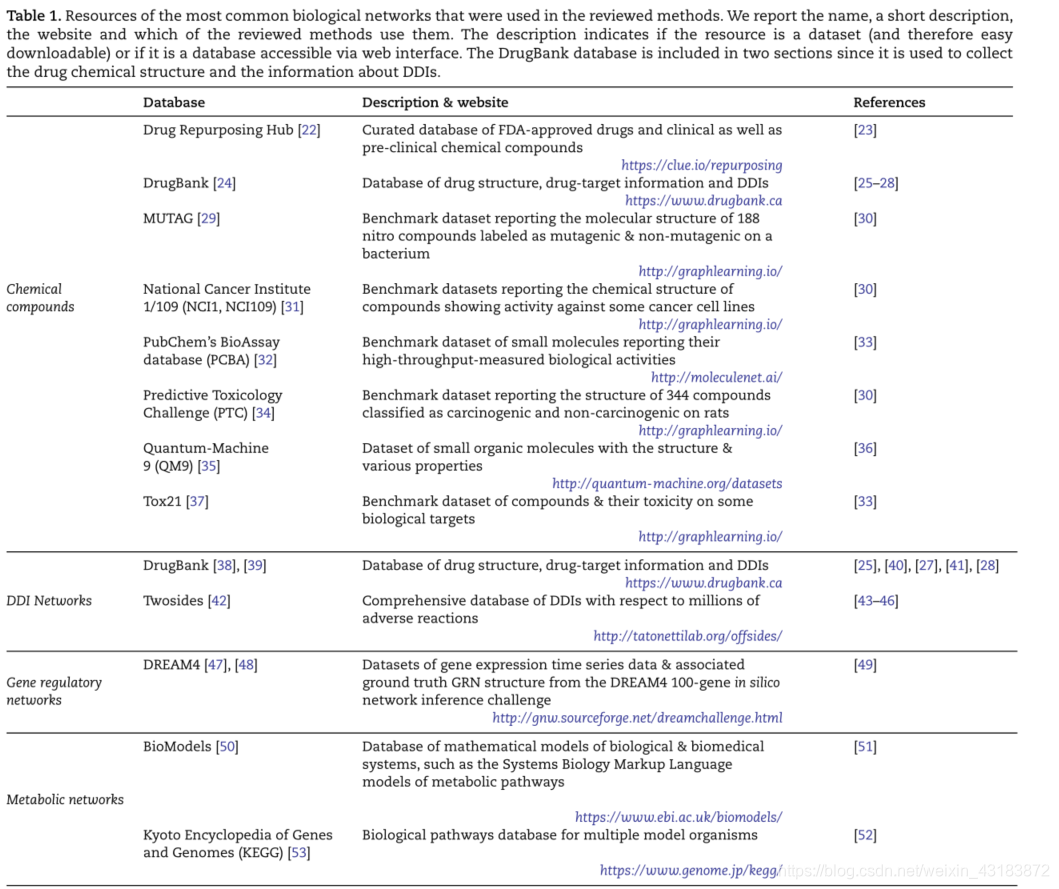
正如我们所看到的,生物网络是表示生物数据的丰富方式,因为它们不仅捕获关于实体本身的信息,还捕获关于这些实体之间关系的信息。关于这些网络的大量信息已经可用,我们在表1中报告了一些在审查方法中使用的最相关的生物网络资源。除了是生物过程的有效表示,生物网络还开启了一套从图形数据中获取新见解的方法。在下一节中,我们将介绍可以在这种图形结构数据上表述的经典类型的问题。

2. Learning tasks on graphs
关于图的学习任务在较高层次上分为节点分类、链接预测、图分类和图嵌入,尽管正如我们将讨论的,为一个任务设计的方法通常可以适用于解决多个任务。我们现在将更详细地解释每个任务。
Node Classification 节点分类生物网络分析中的一个典型任务是根据蛋白质相互作用网络中邻居的功能来预测蛋白质的未知功能。这个问题被称为节点分类[77],当输入图包含一些带标签的节点,但许多没有标签时,这个问题很重要,目标是对网络中剩余的未标签节点进行分类。这通常通过某种形式的半监督学习来解决,其中算法在训练期间使用整个网络作为输入,目标是对所有节点进行分类。虽然所有节点都将被分类,但是在训练期间,仅在具有真实标签的节点上计算损失,从而从具有标签的节点中学习,以便对剩余的未标记的节点进行分类。
Link Prediction链接预测目前关于生物网络中相互作用的知识通常是不完整的,例如哪些基因在基因调控网络中调节另一个基因的表达。预测这些缺失的边,即链接预测[78],在处理这种数据时是一项常见的任务,因为它可以用来预测图中的其他边,或者在加权图的情况下,预测边权重本身。这通常也是一个半监督学习问题,其中图中的已知链接用于预测哪里可能存在额外的链接,类似于节点分类设置。或者,链接预测也可以被构造为监督学习问题,其中在为节点学习嵌入之后,训练次级模型来预测给定节点对之间是否存在链接。
Graph Classification or Regression 图形分类或回归当生物网络数据由多个单独的网络组成时,例如分子的3D结构数据集,目标变成预测每个网络的属性,例如分子的溶解度或毒性。这个任务被称为图形分类[79],它将图形数据集作为输入,然后对每个单独的图形执行分类(或回归)。这是最常见的监督学习问题。
Graph Embedding 图嵌入[80–82]是一种寻找图的低维、固定大小的向量表示的方法,例如PPI网络,或者网络中的元素,例如蛋白质。这通常是通过无监督学习实现的。给定将节点或图形表示为固定大小的向量的有用性,这使得图形能够使用任何现成的机器学习算法,在对特定任务使用标准机器学习算法之前,学习图形嵌入通常被用作预处理步骤。
如上所述,生物数据的图形表示使得许多经典学习任务的制定成为可能。虽然今天可用的高通量技术已经产生了大量这样的数据,但它进一步强调了需要新的计算方法来处理和分析它。在给定数据量的情况下,这些方法需要既高效又高效,以便有效地替代以前的方法。深度学习可以满足这两种需求:它为耗时的任务提供了可扩展性,并具有强大的分类性能潜力,其他领域的强大性能提升就是证明。在下一节中,我们将讨论生物网络上使用的深度学习方法背后的原理和基本算法。
二、Graph neural networks
深度学习方法对矢量数据进行操作,由于图形数据不能直接转换为矢量,因此需要特殊的方法来调整深度学习方法以使用图形。
神经网络是一类这样的方法,它使神经网络方法适应图形领域的工作[83]。虽然GNNs领域包含许多不同的子架构,如recurrent GNNs [84,85],spatial-temporal GNNs [86,87] graph autoencoders [83],但我们在此关注目前用于生物网络分析的那些,即图形嵌入技术[80–82] and grah convolutIonal网络(GCNs) [83]。我们注意到,虽然图形嵌入技术与神经网络密切相关,但并不总是被认为是神经网络的子集。然而,网络嵌入是密切相关的,它经常被用作本文提到的深度学习应用程序的构建块之一,因此我们将在GNNs的伞式分类下描述它。在这一节中,我们将首先介绍使用图形时使用的关键符号,并介绍生物信息学中使用的基本图嵌入和GCN算法。
1. Notation
我们将把图G=(V,E)称为顶点集V,其中|V| = n,边集E,eij∈E表示vi和vj之间的边。每一个图G可以用它的邻接矩阵A ∈ Rn×n来表示,如果图是未加权无向的,那么任何边eij在Aij 和Aji处都用1来表示。带有节点属性的图将这些值存储在一个附加的矩阵中,矩阵的维数是节点属性的维数。虽然本节主要讨论同构、非加权和无向图,但值得注意的是图表示的多样性。图可以是异构的,这意味着它们的节点或边可以有多种类型,例如在知识图中[88]。如果G是一个加权图,边eij在A的条目将是边权重,如果G是一个有向图,边eij并不意味着边eji,这意味着A不一定是对称的。
2. Fundamental algorithms for deep learning on graphs
我们现在将详细介绍今天在生物信息学中广泛使用的两个子领域:图嵌入和GCNs,它们除了是生物信息学中最广泛使用的体系结构之外,还是许多其他GNN体系结构的基本构件。我们将提出的算法可以用来解决引言中提出的学习任务,即节点分类、链接预测、图分类/回归和图嵌入。
Graph embedding
虽然图嵌入通常不被严格认为是GNNs的子集,但它与GNNs交织在一起,并且考虑到它对其他GNNs和生物信息学的重要性,这里将详细考虑。图嵌入方法寻求学习图形或图形元素的低维向量表示,例如它的节点。然后,这种嵌入通常被重新用于节点或图形分类,或链接预测任务。
虽然有许多方法可以解决图形嵌入问题,但最具代表性的是DeepWalk [89],node2vec[54]和LINE[90]。DeepWalk[89]利用自然语言处理中的word2vec [91]框架,通过从每个节点生成多个随机行走,然后优化Skip-gram目标函数,来学习图中每个节点的嵌入。Skip-gram训练目标学习一个节点的嵌入,使得它在随机行走中最大化预测其周围节点的概率,就像word2vec学习一个可以预测周围上下文单词的单词嵌入一样。更具体地说,这可以等价地形式化为下面等式中的最小化问题。[89]中的eq2:

其中 v ∈ V→R |V|×d将每个顶点映射到一个d维空间,得到一个大小为| V |×d的矩阵,其中w是节点vi周围上下文窗口的大小。node2vec [54] 扩展通过引入参数来控制随机漫步是偏向深度优先搜索还是广度优先搜索,从而改进DeepWalk引入的框架。LINE[90]采取不同的方法。它寻求学习一种低维嵌入,使得节点的一阶和二阶接近度表示节点是否直接连接以及它们是否分别拥有共同的邻居,都被保留了下来。也就是说,由一条边连接的节点,或者具有相似邻居集的节点,应该在嵌入空间中彼此靠近。通过异步随机梯度下降最小化捕获一阶和二阶邻近的目标函数来训练LINE。一旦学习了节点或图形的嵌入,就可以使用节点对作为输入,以便预测它们之间是否存在链接,例如在node2vec中所做的那样。

Graph convolutional networks
GCNs是GNNs的一个子集,它采用了非常成功的卷积神经网络(CNN)架构[92],适用于图形结构数据。尽管经常与图像一起使用的神经网络能够利用图像中捕获的空间信息和关系,但由于一组图像可以在相同的规则网格上定义,因此图的邻接矩阵的顺序是任意的,因此不能直接转换到神经网络框架。GCN方法在图形上定义并使用基于谱或空间的卷积,提供了类似于中枢神经系统中图像卷积的图形域。
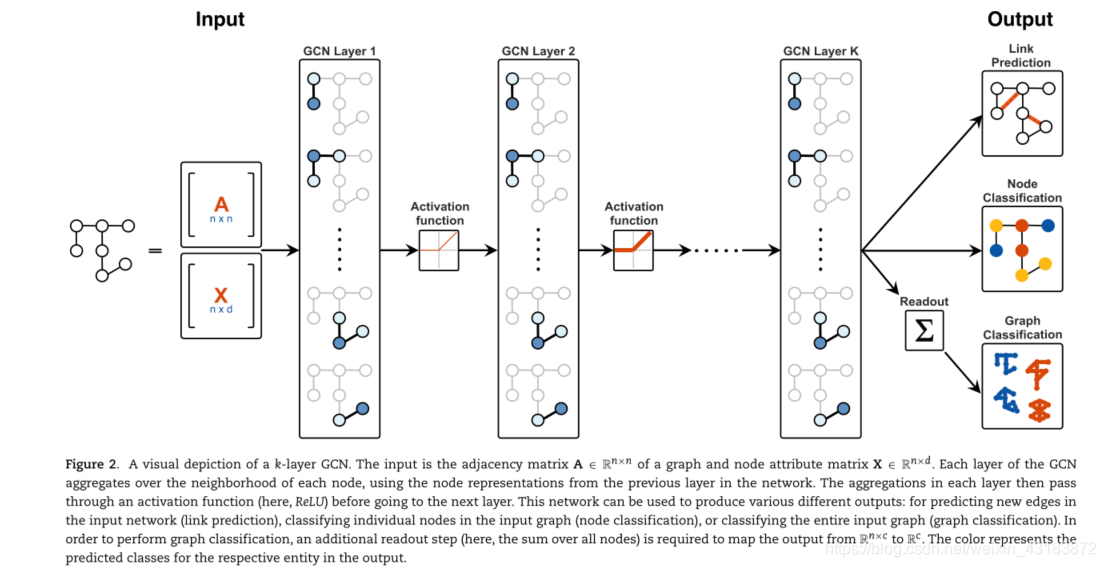
频谱方法,首先由布鲁纳等人[93]引入,后来由德费拉德等人[94]引入,通过使用拉普拉斯图创建傅立叶域中定义的频谱滤波器来构建卷积。然而,由于谱方法所必需的图拉普拉斯特征分解的计算复杂性,使用空间方法开发了更多的方法,其中的思想是通过聚集网络中每个连续层中的邻域来学习每个节点的嵌入。通过对聚合步骤使用排列不变函数,例如和或平均值,可以避免邻接矩阵的任意排序问题,这是阻止图使用标准CNN的原因。每个额外的层包含来自更远的邻域的信息;网络中的k层对应于包含给定节点的k跳邻域。Duvenaud等人[95]就是一个早期的例子,他提供了一个排列不变的卷积,在图中的所有节点上进行运算,并在此过程中计算出一个节点及其邻居的特征之和。虽然最初的设计是检索图形的固定大小的矢量表示,即图形的嵌入,但实际的方法是在图形回归任务上训练的。
Kipf和Welling [96]提供了另一种基于空间的方法,可能是最具开创性的GCNs网络的例子,通常被认为是GCNs网络的基线例子。其重要性还部分归因于这样一个事实,即它们通过展示空间方法的光谱动机来弥合空间方法和光谱方法之间的差距。虽然这种方法最初是作为一种通过半监督学习进行节点分类的方法提出的,但它可以很容易地推广到对图中的高阶结构、边级结果或图本身进行分类。它们为网络定义了一个传播层,其中每一层都有效地结合了来自该节点的k-hop邻域的信息以及节点特征。然后,两层网络的这种前向传播采用下面的形式,由等式Eq 2概括[96]:
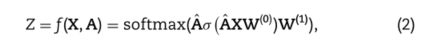
其中,A∈Rn×n是由原邻接矩阵A导出的具有附加自环的归一化邻接矩阵,X ∈ Rn×d是包含所有n个节点的节点属性的特征矩阵,W(i)是来自I层的权重,σ是元素激活函数,如ReLU = max(,0)。在这个例子中,模型的输出Z表示每个节点的类概率,因此Z ∈ Rn×c,c是类的数量。如果h是隐藏单元数,那么W(0)∈R d×h and W(1)∈ Rh×c。
汉密尔顿等人[64]在他们的GraphSAGE算法中提出了类似的想法,但目标是学习一种更具普遍性和计算效率的方法来解决问题。然而,最初的目标是节点嵌入,这也是另一个任务的最终目标,例如节点分类或链接预测。他们通过对一个节点的邻居进行采样,而不是对整个邻居进行采样,并通过学习一个聚合函数来实现这种加速,为此他们考虑了均值、最大值和长短期内存聚合函数。
Gilmer等人[36]从消息传递的角度提供了对图卷积的解释,其中每个节点发送和接收来自其邻居的消息,并且这样做能够更新节点状态。在网络的末端有一个读出步骤,将节点状态聚集到适当的输出级别(例如,从节点级别到图形级别)。令人印象深刻的是,Gilmer等人能够将这里提到的许多论文直接翻译成他们的框架,因此他们的神经信息传递已经成为当今GNNs的一个领先范例。此外,他们测试了这种方案的各种构型,并展示了预测分子性质的最佳构型。
GCNs的这些方法也可以理解为类似于用于测量图形相似性的Weisfeiler–Lehman kernel 的神经网络[97,98],其基于经典的Weisfeiler–Lehman同构测试[99],Kipf和Welling [96]和Hamilton等人[64]明确进行了比较。通过聚集一个节点的所有邻居,使用W的单位矩阵,并将σ设置为适当的哈希函数,可以有效地恢复Weisfeiler–Lehman 算法。因此,GCNs中的适应可以被视为Weisfeiler–Lehman算法和内核的可微和连续的扩展。
在一个完全不同的深入学习图的方法中,尼珀特等人[30]通过对图进行排序来解决节点对应问题,这样做打开了利用更传统的CNN结构的大门。它不是使用完整的图形作为输入,而是为所有图形定义一个公共的固定大小的表示。根据一些预定义的重要性度量,网格中的条目由图中的j个最重要的节点以及j个节点中每一个的k个最近邻居来填充。也可以包括与所讨论的节点相关联的任何对应的节点和/或边缘属性。这样,不同大小的图形都被标准化为相同大小的网格,这使得可以使用标准的CNN过滤器进行学习。
在所有这些方法中,训练是通过迭代计算所有相关样本(如带有标签的节点或图形)的特定任务损失函数来完成的。然后,损失通过反向传播在网络中传播回来。计算权重W的梯度,并根据预定义的更新方程相应地调整权重W。
三、Applications in biology
在回顾深度学习在生物网络上的不同应用时,我们遇到了不同程度的网络信息被包括在内。因此,我们必须定义什么是生物网络上的深度学习。从深度学习的角度来看,我们将其定义为基于非线性函数层次的学习方法。因此,这篇综述集中在深度学习方法上,没有总结使用经典机器学习算法的方法,如核方法、支持向量机、随机森林等,尽管我们将讨论新的深度学习方法相对于经典方法的表现。其次,我们必须定义什么是合格的生物网络,因为一些方法可以使用图的特征,而不需要显式地利用图的结构。例如,可以根据蛋白质中氨基酸的节点标记计数来构建特征向量。是否包括这样的例子并不总是简单明了的。我们最终决定将任何明确讨论或从图形属性生成特征的方法作为有效方法。
我们现在将讨论生物网络分析和深度学习的一些主要用例。我们从更多开始既定实践,即蛋白质分析和药物开发与发现。然后,我们将讨论深度学习作为当前方法的竞争替代物正在出现的应用领域,例如疾病诊断和基因调节和代谢网络分析。我们在补充材料的表2中提供了各种方法的实现信息。总的来说,已经使用经典的交叉验证框架评估了审查方法的性能。有些论文甚至更进一步,使用额外的外部验证数据集来测试所提出方法的可推广性。此外,一些工作甚至通过文献研究或进行实验室实验来验证从头预测。当这两种情况中的任何一种出现时,都会明确提及。
1. Proteomics
蛋白质在许多生物过程中起着关键作用,因此更好地理解它们的作用和相互作用对于回答各种生物问题至关重要。深度学习已经成为回答这些经典问题的一种有前途的新方法。在这一节中,我们将重点关注蛋白质深度学习任务的三个主要类别:预测一对蛋白质是否会相互作用,确定给定蛋白质的功能,以及预测蛋白质的3D结构。
Protein interaction prediction
如引言中所述,PPI网络中的节点是蛋白质,节点之间的边缘代表相互作用。给定一个有代表已知蛋白质相互作用的边的蛋白质图,目标是预测图中的其他蛋白质对也可能相互作用。从图论的角度来看,这是一个链路预测问题。使用GCNs使这些方法能够直接结合网络信息,这通常不包括在经典的机器学习方法中。传统上,许多方法使用氨基酸序列的一级结构来对蛋白质进行载体化和分类。然而,与仅使用序列信息相比,最近利用图结构的方法表现出更强的性能,下面将更详细地讨论。
作为对经典方法的更广泛的评估,岳等人[41]评估了生物信息学任务的其他领域中基于网络的最先进的方法,以提供该领域应该改进的基线性能。这些方法通常将网络嵌入与另一种深度学习方法相结合,以评估其在预测PPI网络中的链接方面的性能,并得出结论,最近基于神经网络的嵌入方法在生物信息学任务中显示出最大的潜力,并且优于传统方法。
刘等人[60]将蛋白质相互作用预测从纯基于序列的载体方法增强到也使用结合网络信息的方法。他们建议通过在蛋白质相互作用指数上使用通用的GCN框架来学习每个节点的表示,该框架编码蛋白质的一级结构序列。每一对蛋白质的表达后来被用作深层神经网络的输入,以预测一对蛋白质是否会相互作用。这种方法扩展了DeepPi[59]的先前工作,该工作使用对蛋白质序列的矢量摘要的深度学习来预测链接。DeepPPI在准确性、精确度和召回率等多种指标上优于SVM、随机森林和朴素贝叶斯等经典方法。刘等人的模型甚至超过了DeepPPI的性能,显示了将网络信息纳入模型的价值。
张和Kabuka [100]试图捕捉蛋白质数据的复杂性,并通过结合数据的多种形式,如一阶和二阶相似性,以及从蛋白质序列中提取的同源性特征,直接使用拓扑特征。他们对数据进行预处理,根据氨基酸组成等特征为每种蛋白质形成一个向量摘要,然后使用无监督和有监督的学习方法相结合来预测相互作用。除了比经典方法(如最近邻法和朴素贝叶斯)具有更好的准确性和精确性之外,他们还表明,他们最先进的预测性能方法在来自八个不同物种的数据集上保持不变。
Protein function prediction
蛋白质分析的另一个领域在于预测蛋白质的功能,因为对高通量实验产生的大量数据进行人工评估相当缓慢且成本高昂。提出这个问题有两种典型的方式:作为节点分类任务或图形分类任务。正如我们将在下面讨论的,这里回顾的新的深度学习方法通常与基于经典机器学习方法的最先进的方法进行比较,并报告其优于它们。
节点分类在节点分类方法中,输入是PPI,其中只有一些节点(即蛋白质)的功能是已知的。任务是对未知节点的功能进行分类。先前讨论的一些预测PPI的方法也被用来对网络中的节点进行分类。例如,在“图形神经网络”一节中描述的两种经典GCN算法,GraphSAGE [64]和Node2vec在PPI数据集上得到验证,并用于预测网络中蛋白质的功能。张和Kabuka预测PPIs的方法[100]也被扩展到给定蛋白质的功能分类。同样,岳等人[41]也评价了各种网络算法在预测蛋白质功能的节点预测任务上的性能。
在一种新的方法中,Gligorijevic 等人[71]考虑使用同一网络的多个表示来表示PPI网络。每个网络包含不同的信息,但使用相同的节点集。他们使用Cao等人[101]的“随机行走并重新启动”来创建每个节点的向量表示,然后为每个邻接矩阵构造一个正的逐点互信息矩阵,该矩阵用作多模式深度自动编码器的输入。该设置允许给出多个PPI作为输入,并促进所有这些信息的整合,最终产生一个低维向量,然后将其提供给SVM用于蛋白质功能分类。作者发现,使用深度学习自动编码器可以学习更丰富、更复杂的网络向量嵌入,与以前基于经典机器学习方法的先进方法相比,性能更好。
曾等人[56]试图从蛋白质相互作用网络中鉴定必需蛋白质。他们使用node2vec [54]学习每个节点的密集向量表示,并将其与使用RNN从基因表达谱中学习的表示相结合。然后通过一个常规的完全连接的网络传递,以便将每个节点分类为必要的或非必要的蛋白。他们再次将他们的方法与经典的机器学习方法(如SVM、决策树和随机森林)进行比较,发现他们的方法在准确性、召回率和AUC等指标上优于所有这些方法。此外,通过一项消融研究,作者揭示了他们驱动性能的方法的最关键的组成部分是PPI的网络嵌入,展示了在网络中捕获的有价值的信息。
OhmNet [65]提供了另一种方法,通过使用从不同组织生成的多层PPI网络,以无监督的方式学习节点的表示。通过整合组织间的差异,这为细胞功能提供了更丰富的信息。该表示是基于网络体系结构学习的,是对node2vec [54]的扩展,用于多尺度图,该图后来用于对网络中的蛋白质功能进行分类。他们将自己与经典方法(如基于张量分解和支持向量机的方法)以及一些基线网络嵌入方法(如line和node2vec)进行了比较,发现在AUROC和AUPRC方面,他们的性能优于所有这些方法。他们将这种优势归因于拥有跨组织蛋白质的多尺度视图,而以前的方法通常将这种视图建模为单个网络。
Graph Classification 第二种方法将蛋白质二级结构元素的图形作为输入,并将其分类为功能组。虽然有许多经典的方法来解决这个问题,如[102],但深度学习提供了一种解决问题的替代方法。在“图形神经网络”一节中提到的几个经典GCN方法使用蛋白质功能预测作为其方法的应用,如尼珀特等人[30]。除了任务是分类而不是回归之外,问题的表述与药物性质预测非常相似,在“药物性质预测”小节中进一步讨论。鉴于强烈的重叠,我们将具体方法的讨论留给该小节。
Protein structure prediction
蛋白质功能预测的一个相关问题是蛋白质结构预测。由于蛋白质的3D结构在很大程度上决定了其功能,这两个问题是相互关联的。最近的工作集中在开发从蛋白质的遗传序列预测其3D结构的方法,也称为蛋白质折叠问题。虽然以前曾努力使用深度学习来预测残基接触,以帮助解决蛋白质折叠问题[103,104],但阿尔法折叠[76]代表了一种突破性的方法,它设定了一个新的基线,大大高于深度学习和传统方法,因此是我们在此详细讨论的唯一文章。像其他方法一样,AlphaFold从氨基酸序列开始,作为预测3D结构的基础。该输入与从蛋白质数据库收集的其他特征信息相结合,并使用CNN来预测所有氨基酸对之间距离的离散概率分布,以及扭转角的概率分布。与以前仅预测两个残基是否通过一个链接连接的方法相比,预测距离及其相应的分布产生了更有信息性和更准确的结果。作者使用距离和扭转角,以及如果预测导致原子重叠的惩罚,来评估他们预测的质量,称为潜力。然后,他们能够执行随机梯度下降,迭代地改进他们的模型。使用这种方法产生了前所未有的结果,并让人们深入了解了深度学习在解决一些最具挑战性的生物信息学问题方面的潜力。
2. Drug development, discovery and polypharmacy
深度学习最近已被用于改进药物发现和开发过程的两个步骤[105],即:(1)筛选数千种化合物,以找到与先前确定的治疗靶点反应的化合物,以及(2)研究潜在候选药物的特性,例如毒性或吸收、分布、代谢和排泄(ADME)。人们对改进筛选步骤很感兴趣,因为这相当费力、昂贵且耗时。本节开始时,我们回顾了一些论文,这些论文提出了深度学习方法,作为当前人工筛选过程(通常称为药物靶点预测)的替代方法。然后,我们总结了以预测药物性质为目标的深度学习方法。随后,我们讨论了越来越多的兴趣,集中在确定哪种药物组合,即所谓的多种药物,可以有效地治疗机制过于复杂的人类疾病,而不能通过使用单一的来治疗[106]。然而,由于药物组合之间的相互作用,这种治疗可能具有不希望的副作用[107]。因此,识别ddi至关重要,而人工识别几乎是不可能的。我们提交的论文试图通过将深度学习方法与分布式数据挖掘网络相结合来解决这个问题。
Drug–target prediction
在确定了治疗相关的靶标(即蛋白质)后,必须正确确定其与不同化合物的相互作用,以表征其结合亲和力或药物靶标相互作用(DTIs)。这种测试过程通常被称为筛选,其结果包括一系列潜在的候选药物,这些药物显示出与目标药物的高结合亲和力。如前所述,人工筛选既昂贵又耗时,因为必须对数千个分子进行筛选才能找到单一药物。深度学习方法试图克服这一局限性,通常使用DDI网络。因此,图深度学习框架内的药物-靶相互作用预测通常被公式化为链接预测问题。基于图形的深度学习方法表明,它们能够有效地解决各种方法中的药物目标预测问题,实现优于先前最先进方法的性能。
这些方法中的一些遵循系统的方法,其中为了解决预测问题,考虑了几个生物网络(PPIs、DDIs)。属于这一类别的一篇有趣的论文来自Manoochehri等人[40],该论文提出了一种编码器-解码器GCN来预测潜在药物和治疗目标之间的相互作用。该方法以由药物、蛋白质、疾病和副作用组成的异构网络为输入,其中节点可以是药物、蛋白质或疾病。当节点通过相互作用类型决定边缘标签的关系连接时,边缘就存在了,例如药物-药物和蛋白质-蛋白质相似性、药物-蛋白质、药物-药物、蛋白质-蛋白质、药物-疾病和药物-蛋白质副作用相互作用。作者结合不同的数据资源来构建这个网络。编码器将所描述的网络作为输入,并返回节点的嵌入,解码器使用该嵌入来捕捉药物-蛋白质相互作用。该过程的输出是蛋白质和药物对之间存在边缘的估计可能性。该方法的稳定性和灵活性是根据异构网络的实际变化来评估的。
曾等人[28]遵循类似的系统方法来解决DTI预测问题,提出了一种称为deepDTnet的方法。[40]和DeepdNet[28]都优于该领域的最新方法。此外,deepDTnet与经典的机器学习方法,即随机森林、支持向量机、k近邻和朴素贝叶斯进行了比较,并在额外的外部验证集上优于它们,证明了它们方法的可推广性。此外,与基线相比,deepDTnet显示出更高的鲁棒性,因为它在显示高和低连通性以及高或低化学相似性的药物或目标上表现良好。deepDTnet的预测在体内实验室实验中得到进一步验证。
另一类方法通过考虑和分析药物和靶标的分子结构来表征DTI。在[26]中,作者提出了一种GCN方法来解决DTI预测问题,其输入由两个图组成,一个蛋白质口袋图和一个2D药物分子图。他们的方法由两个步骤组成,即(I)由用于学习一般口袋特征的自动编码器组成的初步无监督阶段,和(ii)有监督图卷积绑定分类器。后者由两个并行工作的GCN模型组成,即口袋模型和药物GCN模型,分别从蛋白质口袋图和2D分子图中提取特征。有一层负责整合蛋白质之间的相互作用,产生一个联合的药物靶标指纹,然后将其分为“结合”和“非结合”两类。作者将他们的模型与该领域流行的现有深度学习方法和对接程序进行了比较,并报告了更好的性能。在外部验证数据集上获得的结果显示,与基线相比,[26]具有更高的普遍性。
Fout等人[75]介绍了另一种方法来预测给定的一对蛋白质是否会相互作用,以达到药物靶标预测的目的。在这种方法中,给出两个图形作为输入:配体蛋白和受体蛋白。两个图中的节点都对应于残基,每个节点都连接到k个最近的其他节点,这些节点由它们的原子之间的平均距离决定。这不是简单地预测一对蛋白质是否相互作用,而是预测它将在蛋白质的哪个特定位置相互作用。他们的方法是Duvenaud等人[95]引入的指纹方法的扩展,但是通过训练不同的权重允许中心节点相对于其邻居的不同权重,并且允许包括边缘特征。这种方法优于基于使用SVM的另一种最先进的方法。
最后,PotentialNet 是由范伯格等人[108]提出的一个神经网络家族,它不同于以前的神经网络,因为它除了考虑图形分子结构之外,还考虑了不同分子之间的非共价相互作用作为输入。更具体地说,该方法包括三个阶段:(I)仅在共价键上的图形卷积,(ii)同时考虑原子之间的空间信息的共价和非共价传播,以及(iii)仅在配体原子上进行的图形收集步骤,其表示是从键合的配体信息和与蛋白质原子的空间接近性导出的。[108]中的交叉验证策略特别有趣,因为它通过模拟真实的DTIs预测场景来测试潜在网络的泛化能力,例如预测未知分子的亲和力特性。此外,潜在神经网络在分子亲和预测领域可与经典的机器学习方法相媲美。
End-to-End Drug Discovery & Developmen 虽然上述方法仅解决了筛选步骤,但斯托克斯等人[23]最近引入了一种方法来解决整个药物发现和目标验证步骤。受抗生素抗性细菌显著增加和发现新抗生素的困难的驱使,作者提出了一种深入学习的方法来识别对目标细菌即大肠杆菌具有生长抑制作用的分子,他们研究的方向是发现其分子结构不同于现有已知抗生素的候选药物。与其他药物目标预测方法不同,这是一个图形分类问题。一种有向信息传递神经网络,命名为Chempur[109],它是一种根据分子对大肠杆菌的作用进行标记的特征丰富的图形表示。由于前一步主要捕获局部属性,因此也给分类器提供了全局特征分子表示[110]。在学习步骤之后,将获得的分类器部署在几个包含超过1.07亿个分子的化学文库上,以获得可能对大肠杆菌具有抗菌性的潜在候选化合物的列表。然后,根据研究的临床阶段和惩罚与训练分子的相似性和毒性的预定义分数,过滤识别的分子。这一程序导致从药物再利用中心鉴定出了盐霉素。对盐霉素的性质和作用机制进行了实验研究,结果证明其对大肠杆菌和小鼠体内其他细菌具有抗菌活性,表明深度学习可以更及时、更经济地有效改善抗生素发现筛选过程。
Prediction of drug properties
筛选步骤提供了一系列与治疗目标具有高亲和力的分子,在筛选步骤之后,必须对这些候选分子的性质进行研究。这就变成了图分类或者回归问题。我们将回顾寻求预测这些性质的方法,例如以图形表示的化合物的吸收、分布、代谢和排泄(ADME)、稳定性、溶解度、毒性和量子性质。以下方法与经典的机器学习方法进行了比较,结果具有竞争力,详情如下。这一事实突出了深度学习从图形结构中获取有意义信息的有效性,因此它有可能为预测药物特性的经典最新方法提供替代方法。
ADME预测是Chemi-Net [111]的目标,Chemi-Net[111]是一种将GCN和多任务深度神经网络相结合的方法,可以同时解决多个学习任务。Chemi-Net的输入是由两个特征集表示的分子,分别描述原子和原子对。第一个操作包括将原子和原子对描述符的集合投影到三维空间,以获得分子形状的图形结构。后者经过一系列图形卷积运算,然后在读出步骤中将其输出减少到单个固定大小的分子嵌入。在最后一个嵌入表示通过几个完全连接的层之后,获得ADME预测。作者比较了通过使用GCN的嵌入,例如单任务学习化学网络,以及使用传统属性描述符时实现的嵌入。Chemi-Net在几乎所有数据集上都优于基线,除了小的有噪声的数据集。作者通过多任务学习框架克服了这一限制,该框架允许他们利用包含在大数据集内的信息来补偿小数据集。
稳定性是药物发现和开发过程中需要研究的另一个重要特性。DeepChemtable[112]中提出的方法旨在通过结合GCN机制和注意机制,从化合物的图形表示中预测化合物的稳定性。GCN能够在局部水平捕捉分子结构,而注意力机制学习全局图形信息。DeepChemStable研究哪些特征会导致化合物的不稳定性,这使得它能够获得更多可解释的结果。作者将DeepChemStable与基于朴素贝叶斯的基线进行了对比,展示了所提出的深度学习框架的潜力。DeepChemStable和基线在AUC和精度方面具有可比性,而DeepChemStable在召回率方面更胜一筹。潜在的神经网络[108],在“药物靶标预测”小节中介绍,用于差热分析预测,在药物分子性质预测中有进一步的应用,其性能也具有竞争力或优于现有方法。
此外,一些基本的GCN算法试图解决药物性质预测的问题。如前所述,Duvenaud等人[95]提出神经网络的方法来寻找每个分子的指纹,然后将其用于预测分子的药物特性,如分子的溶解度、药物功效和有机光伏效率,并显示出相对于最先进的圆形指纹方法的改进性能。科尔斯等人[33]通过对除了节点信息之外的边缘信息进行卷积,扩展了这一思想。Niepert等人[30]提出的PatchySan算法(也在前面讨论过)也用于根据分子的致癌性对其进行分类[30],并发现其分类精度与经典的基于核的方法相似或更好。最后,如前所述,Gilmer等人[36]迭代现有的GNN方法(重新定义为消息传递),在现有的深度学习方法中找到预测分子性质的最佳配置。
DDI prediction
如前所述,在复杂疾病的情况下,多药疗法是一种有前途的治疗方法,但有一个成本:共同给药的药物之间可能存在不良相互作用,即多药副作用。同时接受多种药物治疗的多种疾病患者经常报告出现副作用。由于实验室筛选药物直接给药系统非常具有挑战性且费用昂贵,因此人们对使用计算方法研究和预测药物相互作用越来越感兴趣。因此,本节将回顾一些使用生物网络来预测药物之间相互作用的深度学习方法,这通常被表述为链接预测问题。如下所述,综述的基于图的深度学习方法通常在很大程度上优于用作基线的经典机器学习和深度学习方法,这表明基于图的深度学习方法可以捕获对DDIs预测问题的有意义的见解。
Decagon [46]是一种用于多关系链接预测的创新GCN方法,它在大型多峰图上运行,其中节点,即蛋白质和药物,根据相互作用类型通过不同种类的边连接。这些多模式网络是结合质子泵抑制剂、顺铂和药物-蛋白质相互作用网络构建的。一旦获得多模态网络,Decagon执行两个主要步骤:编码和解码过程。第一步由一个GCN执行,它接收图并给出它的节点嵌入。第二步由张量分解解码器执行,该解码器从作为输入给出的节点的嵌入中获得多相副作用模型。Decagon的主要优势之一是它不仅能够识别药物之间的相互作用,而且能够识别哪种类型的药物。Decagon的表现优于最先进的基线,例如链接预测的经典机器学习方法、图形表示学习方法和多关系张量分解方法,平均高出20%,在某些情况下甚至高达69%。此外,作者还指出了将PPIs网络纳入此类分析的重要性。事实上,68%的药物组合没有共同的靶点,这表明PPI信息可能是理解哪些特定的靶点药物与蛋白质相互作用的关键环节。
在[27]中提出了另一种用于多关系链接预测的编码器-解码器方法。所提出的HLP方法被设计成在ddi的多图表示上执行,ddi被定义为在节点对之间具有作为节点的药物和作为边的多个交互的网络。使HLP成为一种有趣的方法的特征是它除了捕捉局部邻域信息之外,还能够捕捉全局图结构。与类似的多链路预测模型和迪卡侬[46]相比,HLP表现出更高的性能。然而,Decagon是为研究由药物和蛋白质之间的关系组成的网络而定制的,根据Decagon的作者,这一点很重要,而HLP只在DDI网络上工作和测试。
马等人[43]提出了另一种用于DDI预测的方法,该方法通过整合多个信息源并使用注意机制来学习与每个视图相关联的适当权重,从而产生可解释的药物相似性度量。他们使用GCN架构来构建一个自动编码器,一个GCN作为编码器,另一个GCN作为解码器。每种药物都是其图形中的一个节点,但它包含多个具有相同节点的图形,图形每个视图中的边对应于该视图中节点特征之间的相似性。最终,他们希望获得图中每个节点的节点嵌入,并恢复捕获跨视图信息的单个邻接矩阵,这可以预测药物之间的相互作用。将马等人的方法与最近邻法、标签传播法、多核学习法和[96]中的非概率模型进行了比较。结果表明,对于二进制和多进制预测设置,[43]明显优于基线。
如前所述,DDIs代表了寻找复杂疾病治疗方法的一个有前途的研究方向。因此,除了预测多种药物的副作用之外,目前许多努力都是为了发现多种药物治疗。蒋等人[55]提出了一种预测针对不同癌细胞系协同药物组合的方法。作者将该问题表述为链接预测任务。输入是一个异质网络,通过协同DDI、DTI和PPI网络的组合获得,对于研究中的每种癌细胞系是不同的。这个方法的算法是基于Decagon [46],提出了一个GCN编码器,然后是一个矩阵解码器,以预测药物对之间的协同得分。与、随机森林、弹性网络和基于特征的深度学习方法相比,姜等人[55]提出的方法表现出改进的性能。此外,它可与该领域非常流行的最先进的方法相媲美。最后,作者应用该方法来预测药物的重新组合,并发现其中一些已经在文献中报道为协同抗癌。
另一项研究利用GCN方法进行个性化药物组合预测。GAMENet [44]是一种实现这一目标的方法。GAMENet将采用嵌入网络后接双递归神经网络获得的患者表示与从存储模块获得的网络信息相结合。后者以GCN为基础,从两个网络获取信息,即纵向患者电子健康记录(EHR)的图形表示和直接数据接口网络。
[45]中提出的CompNet是另一种支持医生开具药物组合处方的方法。特别地,EHR数据、处方药物记录和不良药物相互作用网络被用于学习患者和药物信息表示,然后将其组合以获得预测。使用关系GCN构建编码药物信息的模块,称为医学知识图表示模块。GAMENet [44]和CompNet [45]都接受了消融研究,以评估纳入DDIs信息的重要性。在这两种情况下,包含DDI网络都会显著提高性能。此外,GAMENet和CompNet在各种有效性指标上优于几种最先进和经典的机器学习方法,包括F1、Jaccard系数和DDI率。此外,CompNet将其性能与GAMENet的性能进行了对比。CompNet在Jaccard系数、召回率、F1和DDI率方面优于GAMENet,而GAMENet仅在精度方面优于GAMENet。CompNet的作者声称,当目标包括推荐药物组合时,回忆比精确更重要。实际上,这种预测系统代表了医生的支持工具,因此其目标是为医生提供广泛而全面的药物联合给药可能性筛选,而不是提供一个精确但有限的列表。
[25]中介绍了一种不同的处理DDI预测的方法。作者提出了一种通过使用所研究药物的图形表示来增强从文本中提取DDI的方法。这种方法将美国有线电视新闻网在文本药物对上使用的结果与通过在它们的图形分子结构上应用GCN获得的结果连接起来。这种方法的动机是,文献中有许多关于不同药物之间相互作用的信息,但并不总是在DDI数据库中报告,或者在开药时容易获得,同时分子结构包含了对相互作用预测有意义的信息。结果显示[25]具有与深度学习最先进的方法相当的性能,包括曾等人[113]基于 [25],其优于用作基线的经典机器学习方法。此外,在[25]中,包含分子结构的信息在很大程度上增强了基于文本的DDI预测。
3. Disease diagnosis
在过去的几年里,通过深度学习来研究疾病诊断已经引起了研究界的极大兴趣。然而,使用图表的方法,特别是生物网络,是少数。[62]中提出的工作位于这个小的研究领域。作者旨在通过结合光谱聚类和中枢神经系统,从整合了基因表达数据的PPIs网络中预测肺癌。作者尝试了所提出的方法的不同配置,以识别表现最好的一个,并从准确性、精确度和召回率方面评估他们的方法。
此外,Rhe等人[72]提出了另一个在生物网络上进行深度学习的例子,以进行乳腺癌子类型分类。他们的方法整合了一个GCN和一个关系网络,并吸收了一个富含基因表达数据的PPIs网络。利用GCN,他们的方法能够学习局部图信息,而使用RN允许捕捉节点集之间的复杂模式。将GCN输出和RN输出相结合以获得分类结果。将该方法与支持向量机、随机森林、k近邻、多项式和高斯朴素贝叶斯进行了比较,并通过蒙特卡罗交叉验证实验获得了性能。结果表明,所提出的方法在所有使用的指标上都优于基线,表明通过GCN学习PPIs网络特征表示可以显著帮助捕获基因表达数据中的模式。
除了使用引言中描述的生物网络进行疾病诊断之外,还有一些研究使用不同类型的网络,如RNA-疾病关联或通过转换生物医学图像获得的图形,并结合深度学习技术。深度学习如今在疾病诊断研究领域越来越受重视,因此我们在下面的段落中报告了其中的一些方法,以展示该领域有多广泛,尽管使用了传统上不被认为是生物网络的网络。
接下来的两个例子是分别使用RNA-disease和基因-疾病关联网络的应用。张等人[114]提出了一种方法,其输入是表示疾病和RNAs之间关联的图形,称为RNAs-疾病网络。作者使用GCN结合图形注意网络来捕捉输入的全局和局部结构信息,目的是预测RNA-疾病的相关性。相反,韩等人[115]的目标是预测基因与疾病的关联。为了达到这个目的,作者提出了两个GCNs和一个矩阵分解的组合。疾病、基因特征和相似图被赋予两个并行的基因控制网络,它们通过内积组合它们获得的嵌入以获得预测。就用作基线的方法而言,[114]和[115]都显示了它们在从RNAs或遗传病关联网络中捕获有用信息方面的有效性。
除此之外,这一领域的研究主要集中在将生物医学图像转换成图形,然后进行分类。例如,张等人[116]使用基于的分类器从多模态神经图像的图形表示中预测帕金森病。马祖罗等人[117]GCN正致力于核磁共振成像图来预测多发性硬化症。GCNs的使用提高了机器学习和/或深度学习基线的性能[116]和[117],显示了GCNs在图像分析研究领域的潜在改进。
另一个例子是[118],其目的是通过乳房x线照片诊断乳腺癌,只需少量标记样本。他们能够通过基于图的半监督学习为未标记图像创建伪标签,其中每个节点是图像,边缘表示图像之间的相似性。然后,美国有线电视新闻网使用真标签和假标签对单个图像进行训练。该方法在具有深度学习的医学图像分析领域中引入了有价值的贡献,在该领域中,训练有效需要大数据集。具体来说,作者开发了一种策略来克服该领域的一个典型限制:标记的数据点很少。相反,他们使用一种算法,该算法允许在深度学习模型的训练过程中包含未标记的数据。结果显示了该策略的优点,极大地提高了性能。
4. Metabolic networks and GRNs
虽然研究不太广泛,但神经网络也被用于分析代谢和生殖网络。这些早期的研究报告了有希望的结果,表明深度学习捕捉数据中非线性的能力可以积极影响对这些复杂和有意义的生物网络的研究。
代谢网络研究和重建代谢途径是更好地理解生理过程、药物代谢和毒性机制等的一个关键方面。据我们所知,文献中缺乏使用基于图的深度学习方法研究该网络的论文。有可能找到大量旨在分析、建模和重建代谢途径的工作,或者其目标是预测药物代谢的工作,但他们使用经典工具[119]。最近的两篇论文,即[52]和[51],符合我们的复习题目。[52]中的方法旨在通过混合方法预测给定化合物所属的代谢途径。它使用GCN学习给定分子图的形状特征表示,然后将其输入到随机森林中进行分类。作者将他们的方法与几种最先进的机器学习方法进行了比较,展示了使用GCNs作为从所研究分子的图形表示中获取见解的方法的积极影响。此外,作者开发了一种方法来解释GCN提供的化学结构参数方面的特征表示,如直径。
[51]中提出的工作目标是不同的。作者旨在通过利用GNN框架利用代谢途径的图形表示结构来预测代谢途径的动态特性。表示路径的图是使用Petri网建模方法从生物化学路径的系统生物学标记语言模型获得的二分图[120]。作者将提出的方法与预测测试集中多数类的分类器进行了对比,并报告他们的方法总是优于基线。文献[51]中的方法代表了一种计算效率高的替代方法,可替代通常用于评估生化途径动力学特性的繁重的数值和随机模拟。
基因调控网络关于基因调控网络的知识对于深入了解复杂的细胞机制是必不可少的,并且可能有助于识别疾病途径或新的治疗靶点。因此,广义回归神经网络得到了广泛的研究,特别是对推理、验证和重构它们感兴趣。这种研究大多是用经典的方法进行的,而开发的基于图形的深度学习方法的数量相当少,如代谢网络。迄今为止,对大量生物而言,还没有或很难获得精选的全局网数据集[49,121]。由于这个原因,GRNs大多是用无监督的方法分析的[121],因为有监督的技术,特别是深度学习,需要大量注释良好的样本才能有效。此外,GRN推理通常是通过使用来自基因表达数据的信息来完成的,这些数据本质上是有噪声的[122]因此对于训练模型来说并不理想。然而,一些深度学习模型,特别是RNNs,报告了有希望的结果,尽管它们不使用任何类型的图形信息来执行任务。一个例子是[122]中的工作,它通过引入非线性卡尔曼滤波器来提高训练质量,该滤波器非常有效地处理数据中的噪声。
管有上述讨论的局限性,Turki等人[49]提出了一个基于图的深度学习方法的例子。作者使用无监督的方法从基因表达时间序列数据中获得GRN的初步版本,该版本通过清洗算法去噪,然后用于训练不同的有监督的方法来执行基因对之间的链接预测。所提出的数据清理算法是至关重要的,并且可以积极地影响广义回归网络分析领域,因为它提高了广义回归网络数据的质量。更具体地,通过将原始特征投影到使用拉普拉斯核函数计算的特征向量的距离矩阵的特征向量上,获得去噪特征。在清理GRN后使用的监督方法包括支持向量机和深度学习方法,如DNN和深度信念网络。后两种方法优于无监督的最先进的基线,尽管没有优于基于线性SVM的方法。
四、Discussion
基于深度学习在其他领域的成功[7,8],深度学习的前景现在也可以在生物网络分析的许多不同领域看到。我们回顾的方法报告使用经典的机器学习算法一致地匹配或击败了以前的最先进的方法,提供了深度学习的核心优势之一的证据:它强大的经验分类性能。
深度学习的另一个优势是它能够有效地处理大数据集[123],这对经典的机器学习方法来说是一个挑战[123,124]。尽管对具有大量数据的深度学习模型的训练过程不是一项简单的任务,但并行和分布式计算的进步使得训练这些大型深度学习模型成为可能[125,126]。最近GPU计算的突破特别好地满足了神经网络的大量矩阵乘法、高内存要求和易并行性[2,第440页]。
最后,鉴于深度学习是一种基于非线性函数层次结构的学习方法,它能够在没有显式特征工程的情况下检测原始数据中的模式。虽然它不是处理非线性关系的唯一方法,但许多简单的非线性层的组合使它特别擅长于在不同抽象层学习模式[126],从而能够检测更复杂的模式。
虽然深度学习方法很有前途,但也有局限性和许多有待解决的问题。深度学习的主要问题之一是缺乏可解释性。虽然最近在这一领域取得了一些进展[127,128],但是深度学习算法的黑盒性质仍然是一个关键的挑战,特别是在生物信息学中,人们对理解深层学习算法的机制生物过程[129,130]感兴趣。此外,在指导医疗决策的模型环境中,可解释性是至关重要的,在这种环境中,如果对预测过程没有足够的理解,医生和患者通常不太可能相信深度学习模型的输出[127]。
另一个问题是需要大的标记数据集,因为深度神经网络有大量的超参数需要调整。虽然最近技术的进步使得大量数据的收集成为可能,但是生物信息学领域经常受到数据质量问题和缺乏可靠标签的困扰,因为许多数据没有标记[127]。在这种情况下,训练可能很困难,并可能限制生物信息学中深度学习的有效性,这可以在例如GRN分析中看到。此外,并非生物信息学的所有应用领域都可以访问大量数据。例如,在疾病诊断中,数据点可以代表单个患者,因此积累深入学习超越所需的大型数据集可能具有挑战性。此外,对疾病相关数据的访问通常受到隐私限制的限制[131],因此导致该领域数据集的大小有限[132]。在这种较小的数据体系中,通常在标准编程库中可用的经典机器学习方法可以是合适的替代方法[133],例如图核[98,102,134,135]及其实现[136]。
尽管有这些挑战,对图形的深入学习是一个活跃的研究领域,并且已经在各种生物信息学学科中取得了令人兴奋的结果,例如蛋白质组学、药物开发和发现、疾病诊断等,正如我们在这篇综述中看到的那样。因此,我们可以预测生物信息学内外新算法的持续发展,这些算法可以用来分析生物网络。此外,从高通量技术的最新进展中产生的数据量将继续增长,为深入学习解决生物网络分析中的现有问题和新问题提供更多机会。
个人总结
随着最近两年深度学习的发展,使用深度学习来进行生物信息学,即生物网络的研究越来越多。特别是使用图神经网络,比较生物网络归根结底还是图数据。这篇文章主要介绍了在蛋白质,基因调控,疾病诊断领域中使用图神经网络的方法论文,这些论文也证实了所使用的方法是优于传统的机器学习方法的。这篇论文算是第一篇讨论深度学习在生物网络应用的综述类论文。作者算是从分子层面进行详细,如果在有一篇深度学习在RNA-疾病关联预测领域应用的综述类论文,就算完美了,争取以后写一篇。