Guia de uso da ferramenta Spateo (2)
Guia de uso da ferramenta Spateo (1)
Recentemente descobri que Spateo é uma ferramenta mágica que pode realizar segmentação de células. Também contém muitos algoritmos interessantes.
Segmentação de RNA
Neste tutorial, assumiremos que temos apenas imagens de RNA sem qualquer coloração e tentaremos usar o sinal de RNA para identificar células individuais.
Isso é feito através das etapas a seguir.
Identifique o núcleo usando genes de localização nuclear (no nosso caso usaremos os genes Malat1 e Neat1).
Identificação de núcleos extras usando RNA não processado.
[Opcional] Estenda a rotulagem nuclear ao citoplasma.
import spateo as st
import matplotlib.pyplot as plt
st.config.n_threads = 8
%config InlineBackend.print_figure_kwargs = {
'facecolor' : "w"}
%config InlineBackend.figure_format = 'retina'
Baixar dados
Usaremos o conjunto de dados da seção coronal do mouse truncado por Chen et al., 2021.
!wget "https://drive.google.com/uc?export=download&id=18sM-5LmxOgt-3kq4ljtq_EdWHjihvPUx" -nc -O SS200000135TL_D1_all_bin1.txt.gz
Carregue a contagem UMI baixada e as imagens de manchas nucleares em um objeto AnnData. Para efeito de segmentação de células, utilizaremos uma matriz de contagem agregada, onde os obs e vars de AnnData correspondem às coordenadas espaciais X e Y, e cada elemento da matriz contém o número total de UMIs capturados para cada coordenada X e Y. .
adata = st.io.read_bgi_agg(
'SS200000135TL_D1_all_bin1.txt.gz',
gene_agg={
'nuclear': ['Malat1', 'Neat1']} # Add a layer for nuclear-localized genes
)
adata
fig, axes = plt.subplots(ncols=3, figsize=(9, 3), tight_layout=True)
st.pl.imshow(adata, 'nuclear', ax=axes[0], vmax=2, save_show_or_return='return')
st.pl.imshow(adata, 'unspliced', ax=axes[1], vmax=5, save_show_or_return='return')
st.pl.imshow(adata, 'X', ax=axes[2], vmax=10)

Identificação de núcleos celulares usando genes de localização nuclear
Como observamos acima, existem regiões de alta e baixa densidade de RNA. Isto requer dividir a imagem em várias regiões de densidade e depois segmentar cada região separadamente. Caso contrário, o algoritmo pode ser mal calibrado e ser demasiado sensível em regiões ricas em ARN e demasiado rigoroso em regiões esparsas de ARN.
Recomendamos começar primeiro (ou seja, dividir o pixel em muitas regiões de densidade de RNA) e depois mesclar as regiões manualmente.
st.cs.segment_densities(adata, 'nuclear', 50, k=3, dk=3, distance_threshold=3, background=False)
st.pl.contours(adata, 'nuclear_bins', scale=0.15)
st.pl.imshow(adata, 'nuclear_bins', labels=True)
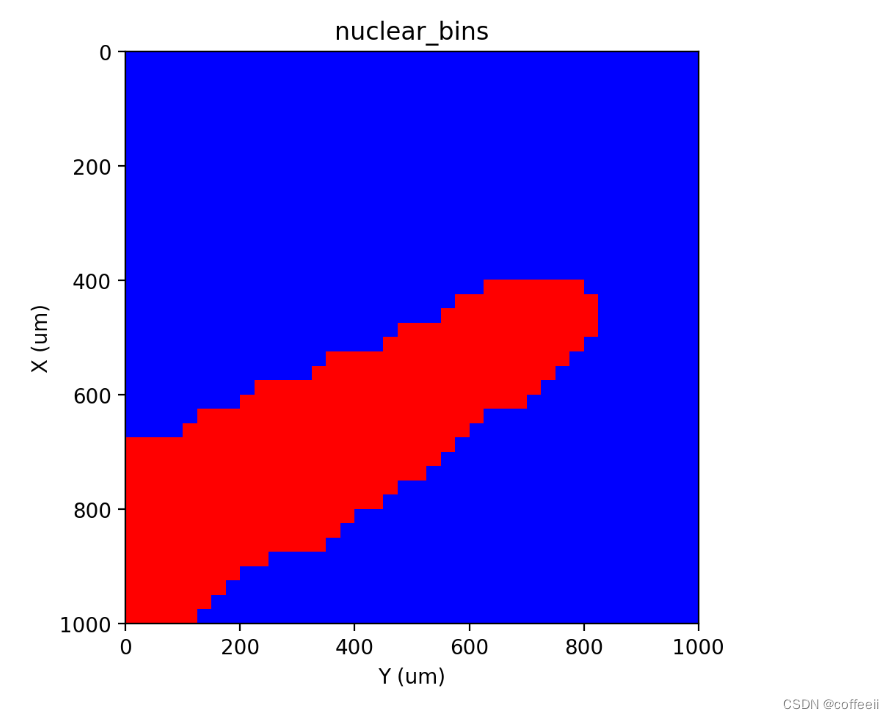
Dividir
st.cs.score_and_mask_pixels(
adata, 'nuclear', k=5, method='VI+BP',
vi_kwargs=dict(downsample=0.1, seed=0, zero_inflated=True)
)
st.pl.imshow(adata, 'nuclear_mask')
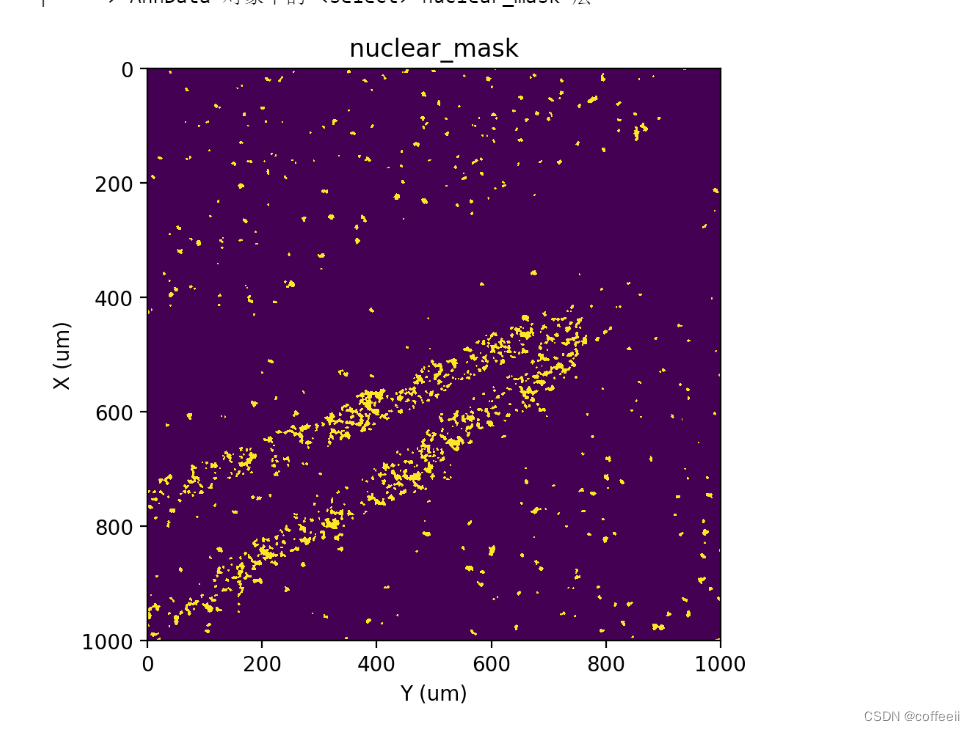
Rótulo
st.cs.find_peaks_from_mask(adata, 'nuclear', 7)
st.cs.watershed(
adata, 'nuclear_distances', 1,
mask_layer='nuclear_mask',
markers_layer='nuclear_markers',
out_layer='nuclear_labels'
)
st.pl.imshow(adata, 'nuclear_labels', labels=True)
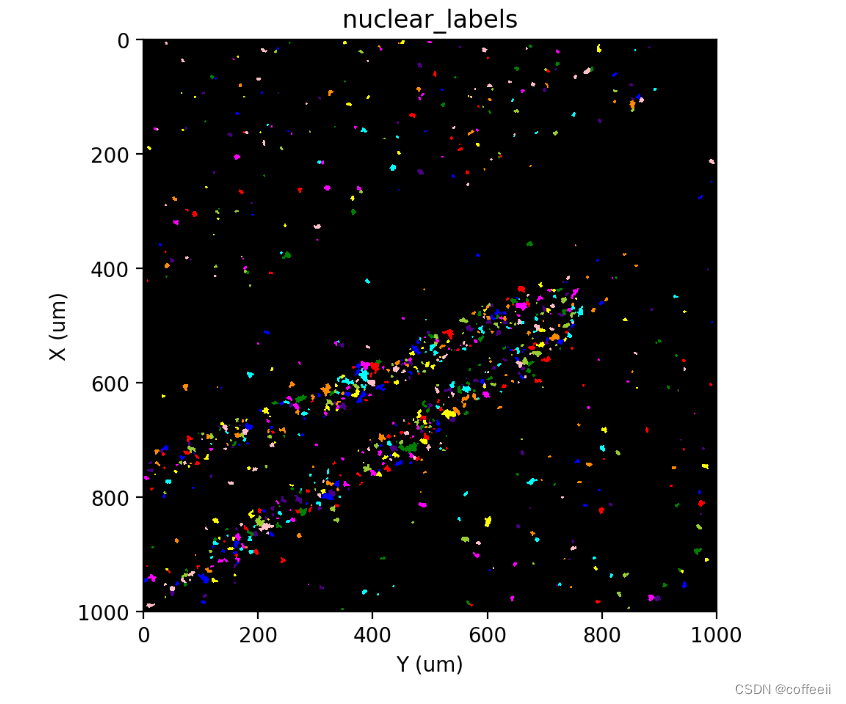
Identificando núcleos extras com RNA não processado
st.cs.segment_densities(adata, 'unspliced', 50, k=3, dk=3, distance_threshold=3, background=False)
st.pl.contours(adata, 'unspliced_bins', scale=0.15)
st.pl.imshow(adata, 'unspliced_bins', labels=True)
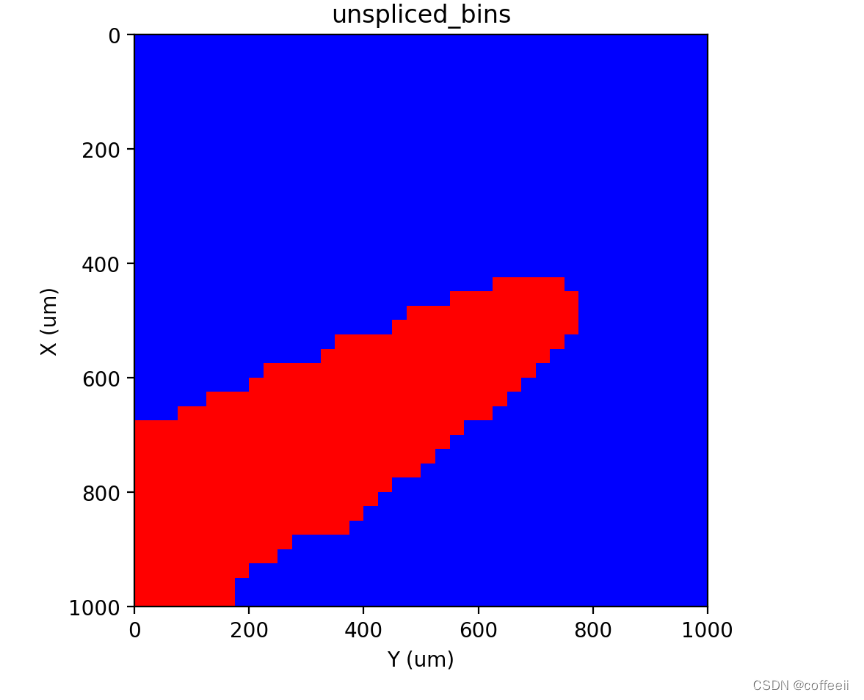
Dividir
Os núcleos foram então identificados usando RNA não processado, como fizemos anteriormente para contagens de genes nucleares. Observe que esta função detectará automaticamente caixas de densidade de RNA e ajustará o algoritmo de acordo. Além disso, observe que fornecemos o parâmetro right_layer st.pp.segmentation.score_and_mask_pixels e o parâmetro seed_layer st.pp.segmentation.label_connected_components para indicar o rótulo nuclear obtido do gene de localização nuclear. Internamente, esses rótulos são usados para auxiliar ainda mais na identificação de núcleos reais.
st.cs.score_and_mask_pixels(
adata, 'unspliced', k=5, method='VI+BP',
vi_kwargs=dict(downsample=0.1, seed=0),
certain_layer='nuclear_labels'
)
st.pl.imshow(adata, 'unspliced_mask')
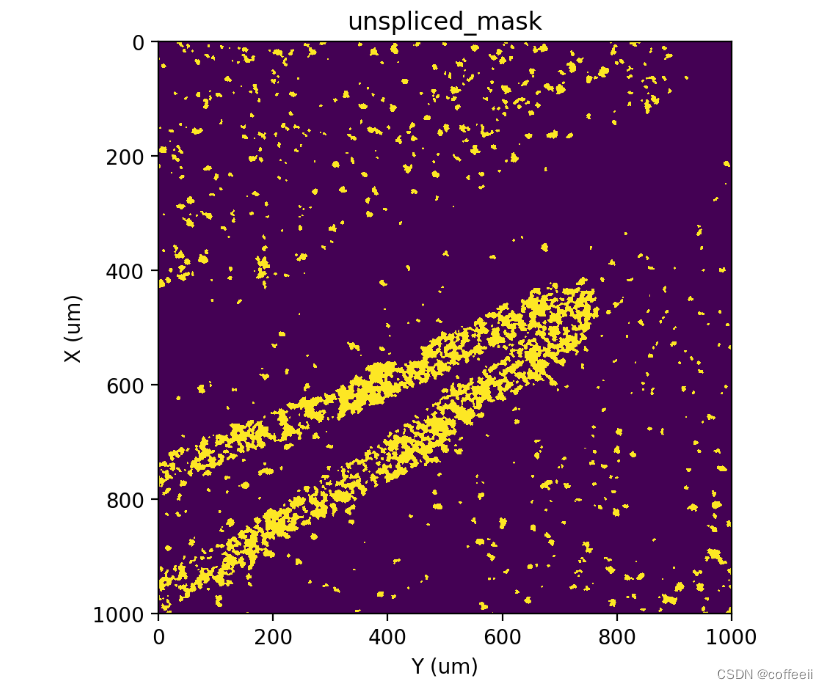
Rótulo
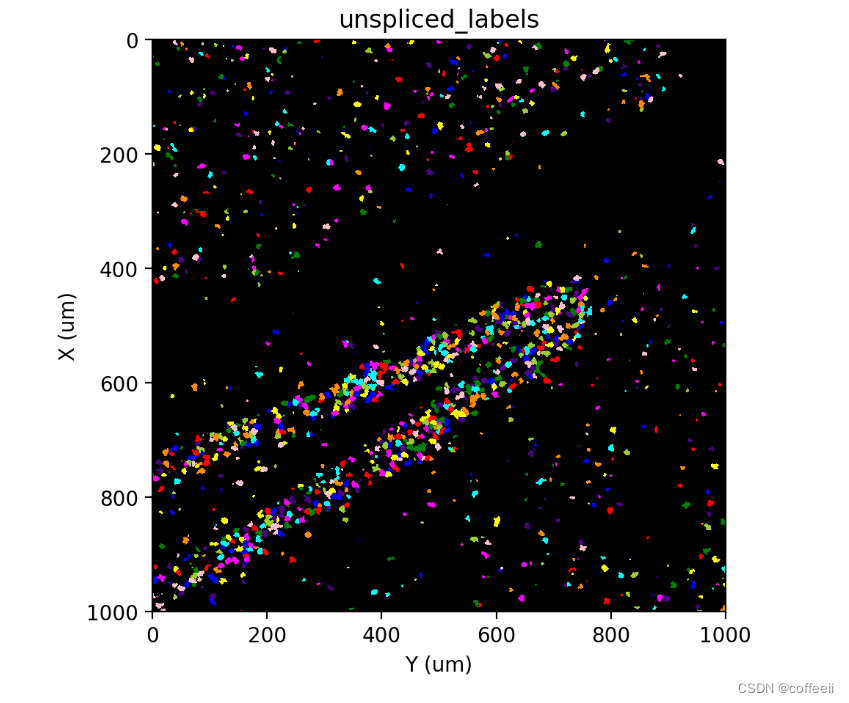
Ao contrário de antes, onde usamos genes nucleares localizados, aqui conhecemos alguns rótulos iniciais que queremos manter (em alguns casos, amplificados para preencher a máscara acima). Além disso, o uso de tags não emendadas parece saturar algumas regiões densas em RNA, tornando difícil discernir os limites das células nessas regiões. Portanto, em vez de usar uma abordagem de divisor de águas que tenta "preencher" a máscara inteira, usamos st.cs.label_connected_components para limitar a área máxima à qual cada rótulo pode ser alocado.
Observe que o parâmetro seed_label que fornecemos é o rótulo que obtivemos anteriormente usando genes nucleares localizados.
st.cs.label_connected_components(adata, 'unspliced', seed_layer='nuclear_labels')
st.pl.imshow(adata, 'unspliced_labels', labels=True)
Para obter mais conhecimento em bioinformática, seja bem-vindo ao intercâmbio v: coffeeiix (treinamento em análise de transcriptoma de célula única também está disponível)