Here, let me introduce the data in the BGEN format. Its file format is as follows: a.bgen, which is a new data format, and its application is not as good as plink's binary files: .bim, .bed, .fam. Here is how to convert each other.
1. Introduction to bgen format
Modern genetic association studies typically use data from tens to hundreds of thousands of individuals genotyped or imputed from tens of millions of markers across the genome. Traditional data formats based on the textual representation of these data (such as the GEN format for IMPUTE output or the variable call format) are sometimes not well suited for these data volumes. In fact, for simple programs, the time spent parsing these formats can dominate program execution time.
This page describes the binary GEN file format (the "BGEN" format), which is designed to address these issues. BGEN is a robust format designed with specific mixing properties that we believe are useful for this type of research. It is aimed at large, potentially genetic datasets. Key features include:
-
The ability to store direct input and input data.
-
Ability to store non-staged genotype and staged haplotype data.
-
File sizes are smaller by using efficient, variable-precision compressed bit representation and compression.
-
The use of per-variable compression makes the format easy to index and catalog.
For example, the figure below shows variant identification data (i.e., genomic position, ID field and allele) time required. Both variants of BGEN defined below are shown.
In the figure below, the X week is the belt of the file, which is converted by log10. The Y coordinate is the processing time, the format of the file:
- zipped gen file
- compressed vcf file
- bgen document
- bed-file
- vcf original file
- gen document
It can be seen that the three formats of bgen1.2, bed, and bgen1.1 have the smallest file size and the shortest processing time. The bed file does not contain as much information as bgen, so the bgen format is recommended here.
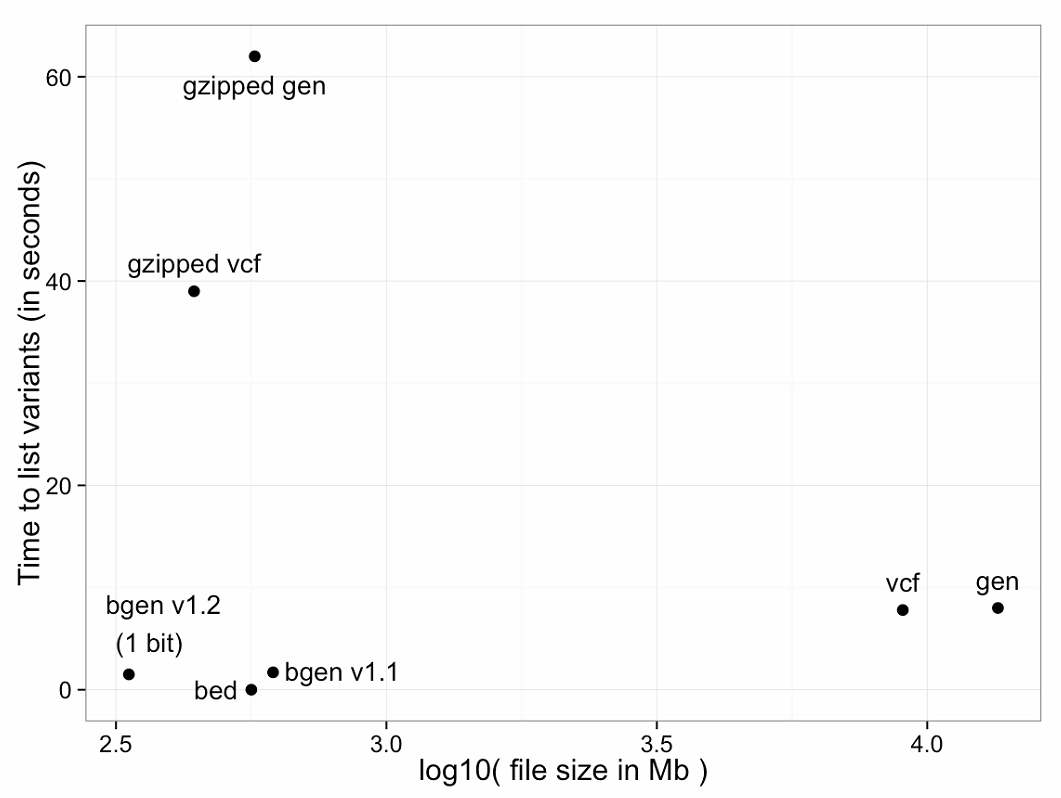
For PLINK binary (.bid) files, the identification data is stored in a separate file (.bim file), so the time is effectively zero. For text-based formats, there is a significant trade-off between the use of file compression and read performance. BGEN stores the entire dataset of 2.25 billion genotypes at 334Mb, a little more than one bit per genotype, and took 1.5 seconds on this test.
(Of course, performance optimizations are possible for all formats, so the graph above will not represent the best possible timing, but should be considered illustrative.)
The BGEN format has been used in several major projects, including the Wellcome Trust Case Control Consortium 2, the MalariaGEN project, and the ALSPAC study. It has been adopted by the UK Biobank as the publishing format for genome-wide estimated genotypes.
2. Software that handles bgen
Here, commonly used software:
- Mega2
- LDstore
- PLINK
- STITCH

For R language users, you can use: rbgenpackage processing
For Python users, you can use: bgen-readerand pybgenpackage processing
There are also some C++ programs that can be processed. For details, please refer to: https://www.well.ox.ac.uk/~gav/bgen_format/software.html
3. Convert bgen format to plink file (ped, map)
Note that when plink reads the bgen file, you need to specify:
- .bgen
- .sample
Both files must exist. The bgen file is a binary file, and the sample file is four columns of data including ID_1, ID_2, and missing sex.
plink2 --bgen t1.bgen 'ref-last' --sample t1.sample --export ped --out x1
- –bgen file: Specify t1.bgen, followed by parameters:
ref-last, indicating that the ref is placed behind, instead of the default major as ref - –sample file, specify t1.sample, followed by sample file, these two files should be specified separately
- –export ped, specify the format of the output, which is a text file for outputting plink
4. Convert bgen format to plink binary files (bed, bim and bed)
plink2 --bgen t1.bgen 'ref-last' --sample t1.sample --make-bed --out x1
The parameters are the same as above, and the --make-bedoutput format is defined by the output bed,bim,fam.
5. Convert plink binary file to bgen format
plink2 --bfile a1 --export bgen-1.1 --out t1
- –bfile, read binary prefix
- –export bgen-1.1 output bgen format file, you can also use bgen-1.2
Just sauce!
Other references: Sad notes for plink2.0 and plink1.9