Руководство по использованию инструмента Spateo (2)
Руководство по использованию инструмента Spateo (1)
Недавно я обнаружил, что Spateo — это волшебный инструмент, который может выполнять сегментацию клеток. В нем также есть много интересных алгоритмов.
Сегментация РНК
В этом уроке мы предположим, что у нас есть только изображения РНК без какого-либо окрашивания, и попытаемся использовать сигнал РНК для идентификации отдельных клеток.
Это достигается посредством следующих шагов.
Определите ядра, используя гены ядерной локализации (в нашем примере мы будем использовать гены Malat1 и Neat1).
Идентификация дополнительных ядер с помощью несплайсированной РНК.
[Необязательно] Распространите ядерную маркировку на цитоплазму.
import spateo as st
import matplotlib.pyplot as plt
st.config.n_threads = 8
%config InlineBackend.print_figure_kwargs = {
'facecolor' : "w"}
%config InlineBackend.figure_format = 'retina'
Загрузить данные
Мы будем использовать набор данных коронального среза мыши, усеченный Chen et al., 2021.
!wget "https://drive.google.com/uc?export=download&id=18sM-5LmxOgt-3kq4ljtq_EdWHjihvPUx" -nc -O SS200000135TL_D1_all_bin1.txt.gz
Загрузите загруженные изображения количества UMI и ядерных пятен в объект AnnData. В целях сегментации ячеек мы будем использовать агрегированную матрицу подсчета, где obs и var AnnData соответствуют пространственным координатам X и Y, а каждый элемент матрицы содержит общее количество координат UMI, захваченных для каждого X и Y. координировать.
adata = st.io.read_bgi_agg(
'SS200000135TL_D1_all_bin1.txt.gz',
gene_agg={
'nuclear': ['Malat1', 'Neat1']} # Add a layer for nuclear-localized genes
)
adata
fig, axes = plt.subplots(ncols=3, figsize=(9, 3), tight_layout=True)
st.pl.imshow(adata, 'nuclear', ax=axes[0], vmax=2, save_show_or_return='return')
st.pl.imshow(adata, 'unspliced', ax=axes[1], vmax=5, save_show_or_return='return')
st.pl.imshow(adata, 'X', ax=axes[2], vmax=10)

Идентификация ядер клеток с использованием генов ядерной локализации
Как мы заметили выше, существуют области с высокой и низкой плотностью РНК. Для этого необходимо разделить изображение на несколько областей плотности, а затем сегментировать каждую область отдельно. В противном случае алгоритм может быть неправильно откалиброван и оказаться слишком чувствительным в регионах, богатых РНК, и слишком строгим в регионах с разреженным РНК.
Мы рекомендуем сначала начать свободно (то есть разделить пиксель на множество областей плотности РНК), а затем объединить области вручную.
st.cs.segment_densities(adata, 'nuclear', 50, k=3, dk=3, distance_threshold=3, background=False)
st.pl.contours(adata, 'nuclear_bins', scale=0.15)
st.pl.imshow(adata, 'nuclear_bins', labels=True)
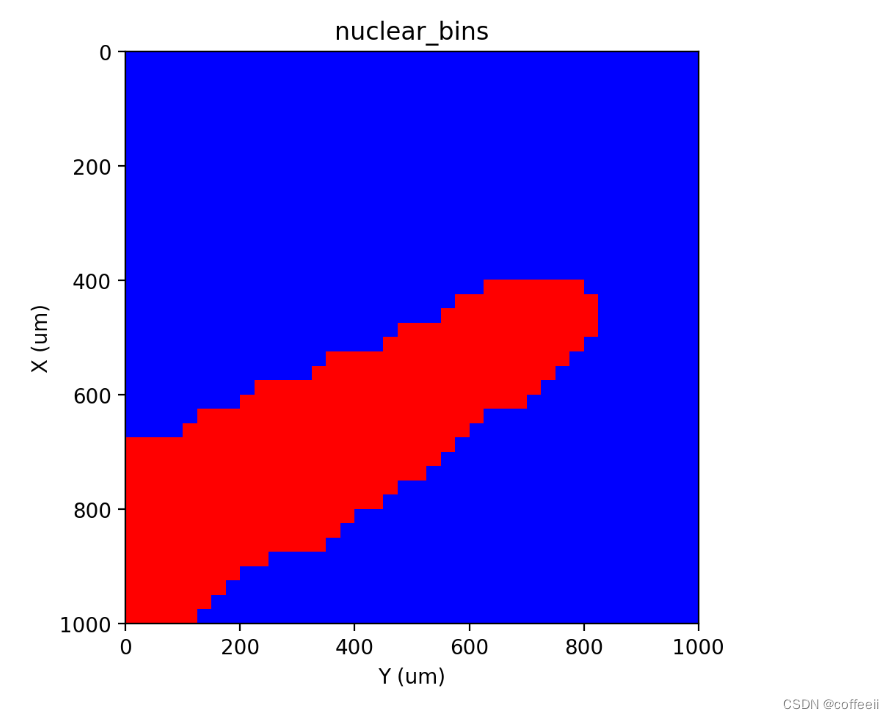
Расколоть
st.cs.score_and_mask_pixels(
adata, 'nuclear', k=5, method='VI+BP',
vi_kwargs=dict(downsample=0.1, seed=0, zero_inflated=True)
)
st.pl.imshow(adata, 'nuclear_mask')
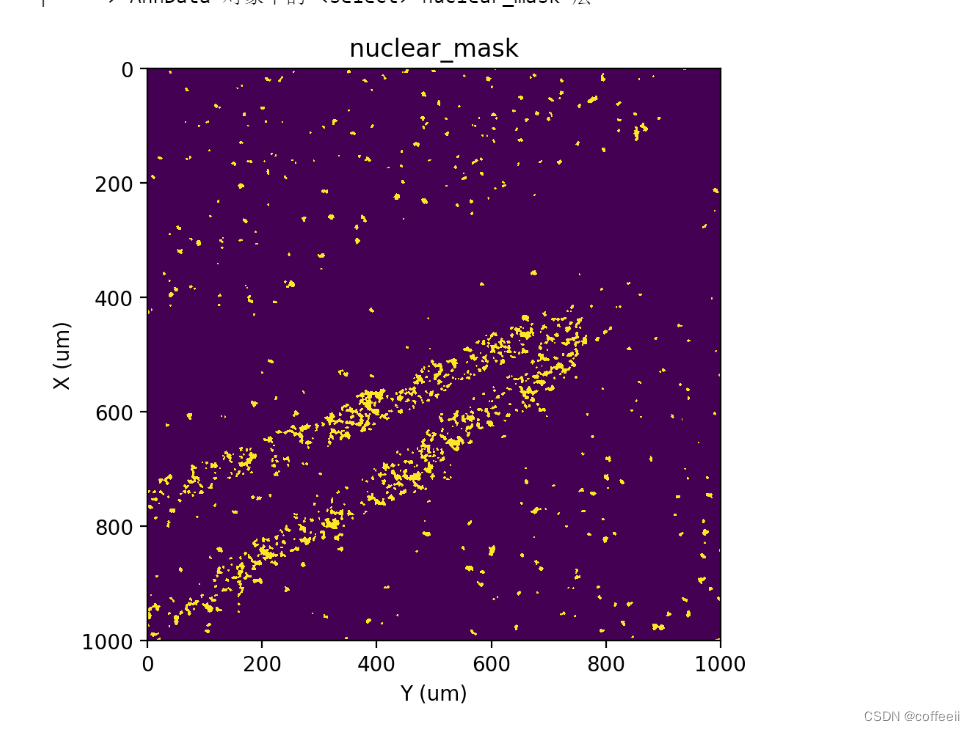
Этикетка
st.cs.find_peaks_from_mask(adata, 'nuclear', 7)
st.cs.watershed(
adata, 'nuclear_distances', 1,
mask_layer='nuclear_mask',
markers_layer='nuclear_markers',
out_layer='nuclear_labels'
)
st.pl.imshow(adata, 'nuclear_labels', labels=True)
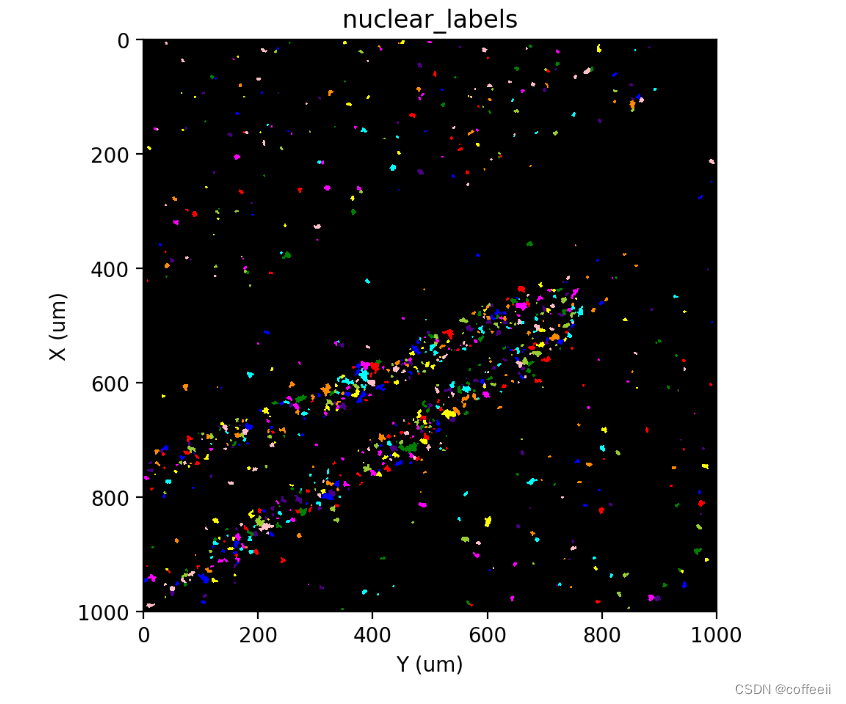
Выявление дополнительных ядер с несплайсированной РНК
st.cs.segment_densities(adata, 'unspliced', 50, k=3, dk=3, distance_threshold=3, background=False)
st.pl.contours(adata, 'unspliced_bins', scale=0.15)
st.pl.imshow(adata, 'unspliced_bins', labels=True)
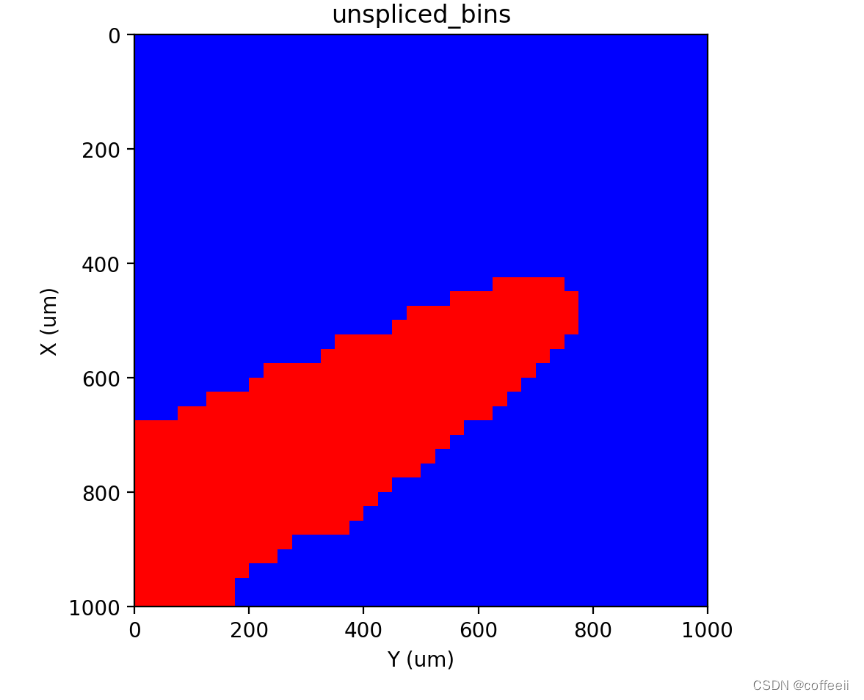
Расколоть
Затем ядра были идентифицированы с использованием несплайсированной РНК, как мы делали ранее для подсчета ядерных генов. Обратите внимание, что эта функция автоматически обнаруживает ячейки плотности РНК и соответствующим образом корректирует алгоритм. Также обратите внимание, что мы предоставляем параметр «определенный_слой» st.pp.segmentation.score_and_mask_pixels и параметр «seed_layer» st.pp.segmentation.label_connected_comComponents для указания ядерных меток, полученных из ядерно локализованных генов. Внутри эти метки используются для дальнейшей идентификации настоящих ядер.
st.cs.score_and_mask_pixels(
adata, 'unspliced', k=5, method='VI+BP',
vi_kwargs=dict(downsample=0.1, seed=0),
certain_layer='nuclear_labels'
)
st.pl.imshow(adata, 'unspliced_mask')
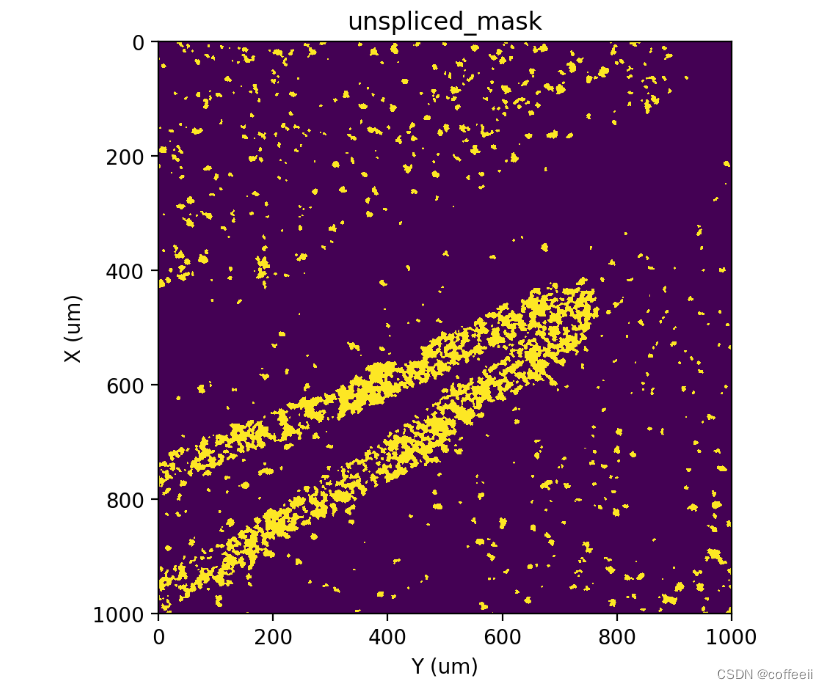
Этикетка
В отличие от предыдущего случая, когда мы использовали ядерные локализованные гены, здесь мы знаем некоторые исходные метки, которые хотим сохранить (и в некоторых случаях увеличить масштаб, чтобы заполнить приведенную выше маску). Более того, использование несплайсированных меток, по-видимому, приводит к перенасыщению некоторых участков с высокой плотностью РНК, что затрудняет различение границ клеток в этих участках. Поэтому вместо использования подхода водораздела, который пытается «заполнить» всю маску, мы используем st.cs.label_connected_comComponents, чтобы ограничить максимальную область, которой может быть назначена каждая метка.
Обратите внимание, что предоставляемый нами параметрseed_label — это метка, которую мы получили ранее с использованием ядерно локализованных генов.
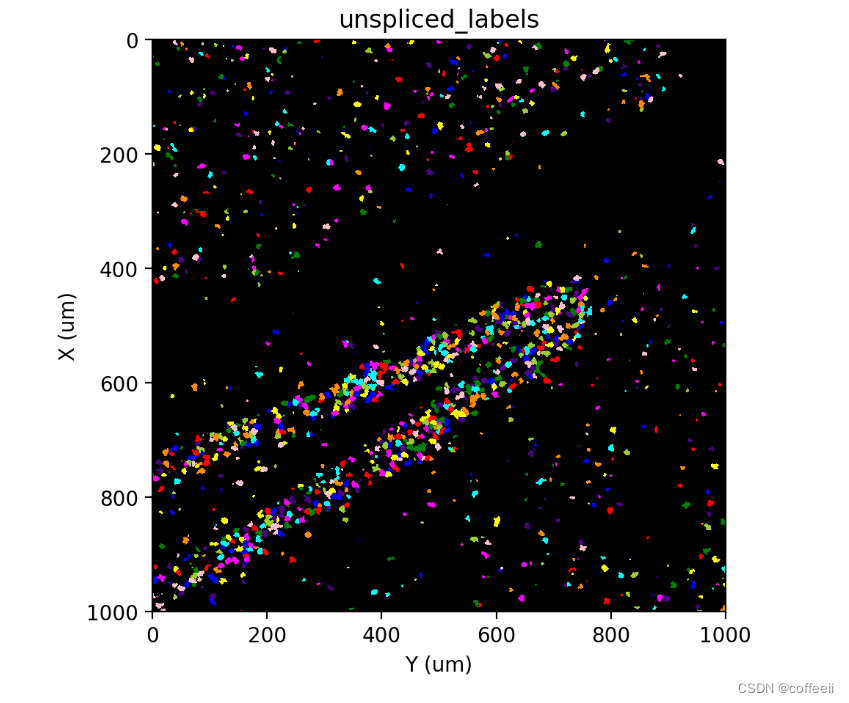
st.cs.label_connected_components(adata, 'unspliced', seed_layer='nuclear_labels')
st.pl.imshow(adata, 'unspliced_labels', labels=True)
Для получения дополнительных знаний в области биоинформатики приглашаем вас на обмен v: Coffeeiix (также доступно обучение по анализу транскриптома отдельных клеток)