1. Introduction to local blast
Local Blast (Basic Local Alignment Search Tool) is a local-based comparison search tool that can perform blast search in a database built by yourself. Compared with NCBI's online blast, it has a faster speed, a smaller search range, and no Internet For example, if you already know a gene of barley, and have clarified its function, now you need to find a gene with high sequence similarity in barley, you can build a database locally, that is, create a database of barley, and then blast to find the same Source sequence.
2. Installation of local blast
1. NCBI download the local version of blast
[Download link]: https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/ to download the version suitable for my computer system, here I download ncbi-blast-2.11.0

2. Install the local blast program
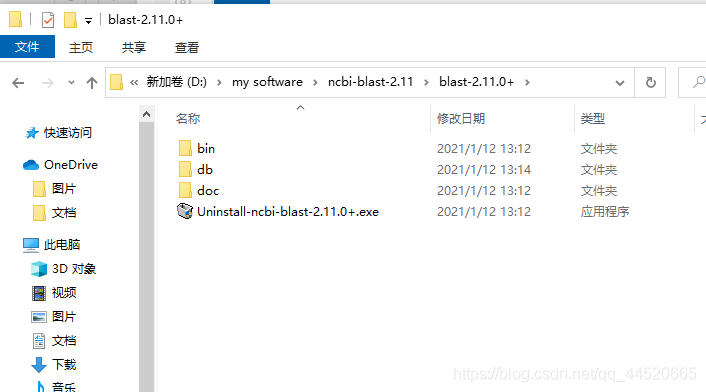
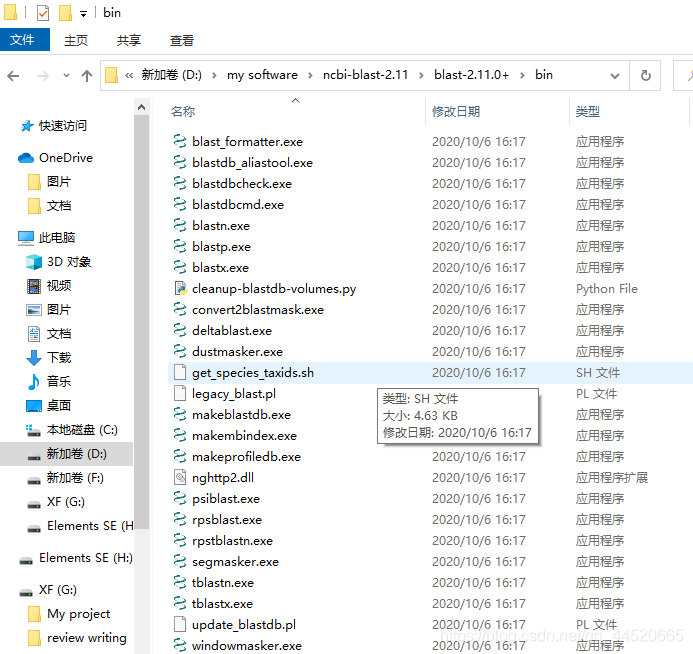
After downloading, double-click to install, you can judge whether the download is complete by comparing the size of the program, generate two subdirectories bin and doc, where bin is the program directory and doc is the document directory, you can also click to check the integrity of the program, and then create a new directory Folder, renamed todb
3. User environment variable settings
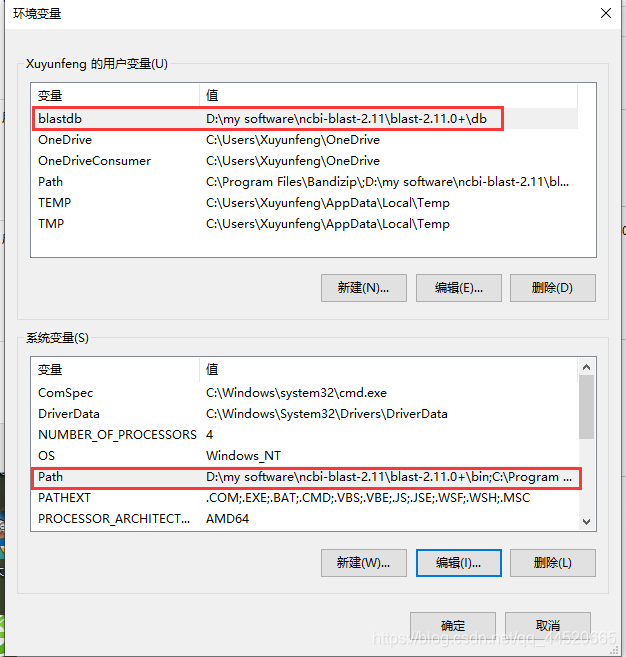
This computer-Properties-Advanced System Settings-Environment Variables
Under User Variables: New-Variable Name: balstdb, the variable value is the path of the newly created db folder after the computer is installed.
Under System Variables: Path-Add variable value to bin on the computer Folder location
3. Build a local blast database
1. Database download
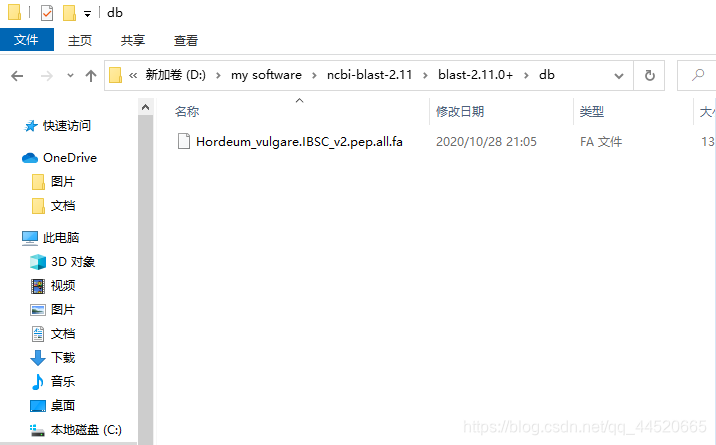
Download barley’s pep data from databases such as ensemble and unzip it todbFolder
2. Database formatting
When no target is selected in the db folder, hold down Shift, right-click on the blank space of the folder, and select Open herePowerShellWindow, open the window as shown in the figure. I use a win10 system to quickly enter the location of the db folder in this way. If it is another version of the windows system, you may need to run cmd to enter the location of the db folder.
attention: The file is in the folder currently opened by Windows powershell, so there is no need to substitute the path.
Run the command:
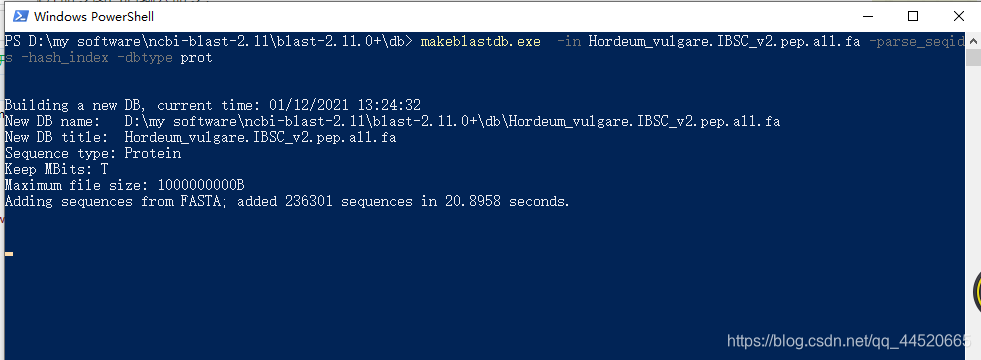
makeblastdb.exe -in Hordeum_vulgare.IBSC_v2.pep.all.fa -parse_seqids -hash_index -dbtype prot
Where Hordeum_vulgare.IBSC_v2.pep.all.fa is the name of the database you want to format, change it according to the name of your own database, remember to add the suffix .fa; prot after dbtype represents the type of database,protRepresents the amino acid sequence database, if it is a nucleotide sequence, usenucl. After the database is formatted in the command line, the following figure is displayed
4. Local blast operation
1. Preparation of query sequence
Created under the folder D:\my software\ncbi-blast-2.11\blast-2.11.0+\dbtarget.seq.txtSave the sequence to be queried into in.txt in fasta format. We have taken a protein sequence of barley as an example

2.Query sequence query
Created under D:\my software\ncbi-blast-2.11\blast-2.11.0+\db folderNew text file.txtThe text file, use the blast command:
blastp.exe -task blastp -query target.seq.txt -db Hordeum_vulgare.IBSC_v2.pep.all.fa -out out.txt -evalue 1e-10 -outfmt 6 -num_threads 2
Related parameter description:
The blastp.exe program executes the command, the program before exe is changed according to your needs, including the programs contained in the bin folder such as blastn, blatp, tblastx;
-After task , select the program you want to use, blastn, blastp, tblastx, etc.;
-query is followed by the file name of the query sequence;
-db followed by the formatted database name;
-out is followed by the name and format of the file to be output. The format includes 0-10, of which 6 and 0 are the most commonly used. You can try it yourself.
-num_threads parameter settings can be set according to the performance of their computers, notebook recommended no more than 2, thereby improving the efficiency ratio to

save after theNew text file.txtRenamed toHordeum_vulgare.pep-blast.cmd, At this time a text file has been modified to the cmd command in windows, double-click to run
blast result description
 Each column indicates:
Each column indicates:
A:Query_id
B:Subject_id
C:Identity
D:Align_length
E:Miss_match
F:Gap
G:Query_start
H:Query_end
I:Subject_start
J:Subject_end
K:E_value
L:Score
E value (Expect) : indicates random match For example, E=1, which means that in a database of the current size, the average number of objects found by chance is 1. The larger the E value, the greater the probability of random matching. When the E value is close to zero or zero, it is essentially a perfect match. Generally speaking, we think that E value less than 10-5 is a more feasible S value result. We can imagine that in the same database, if there are 1000 entries with a chance of S value higher than the current one when E=0.001, then if E is not set to 10-6, only one result may be obtained, that is, the S value is the most reliable. The one. But the E value is not a panacea. It has limitations in the following situations:
1) When the target sequence is too small, the E value will be too large, because a higher S value cannot be obtained.
2) When the homology between the two sequences is high, but there is a large gap (gap), the S value will decrease. At this time gap scores are very useful.
3) When the non-functional regions of some sequences have low randomness, it may cause higher homology between the two sequences.
E-value summary: E-value is suitable for sequences that have a certain length and cannot be too low in complexity. When the E value is less than 10-5, it indicates that the two sequences have higher homology, not because of calculation errors. When the E value is less than 10-6, the homology between the two sequences in the table is very high, and there is almost no need for further confirmation.
Identity (Identities) : or similarity. The number of matched bases is the percentage of the total sequence length.
Score : The higher the score, the better the homology; the smaller the Expectation value, the better the comparison result, indicating that the error caused by some reasons is smaller; Identities are homology (similarity), as shown in the example Only 35 of the 1,299 bases compared are unmatched, and the other 97% are the same;
Gaps : refers to the number of bases that are more or less or missing; deletions or insertions (Gaps): insertions or deletions. Use "—" to indicate.
In addition, the compared Strand is judged by s. Start: and s. End, as in the third row of the above result. The Star value is greater than s. End, it means a negative chain.
Summary and supplement: another method
For local blast sequence comparison, we need to create a library file (the online blast library uses files from major biological databases), and enter the command:makeblastdbBuild database command.
makeblastdb -in b.fasta -dbtype nucl -out b.fasta.blastdb
makeblastdb -in b.fasta -dbtype nucl -out b.fasta.blastdb [The file is in the folder currently opened by Windows powershell, so there is no need to enter the path]
== Note that the space must not be less =
-in is the build library file, we use Larger file build library
-out is followed by the output library file name. Generally, after the first step of building the library, three files nhr/nin/nsq will be generated together as a library to enter the next step.
If you are running a nucleic acid sequence, use blastn
blastn -query a.fasta -db b.fasta.blastdb -out b.blast -outfmt 6 -evalue 1e-5 -num_threads 2
blastn -query a.fasta -db b.fasta.blastdb -out b.blast -outfmt 6 -evalue 1e-5 -num_threads 2
-query : input file path and file name
-out : output file path and file name
-db : Formatted database path and database name
-outfmt : output file format, there are a total of 12 formats, 6 is the tabular format corresponding to the BLAST m8 format
-evalue : set the e-value value of the output result
-num_descriptions : the tabular format output result Number-
num_threads : number of threads [notebook generally does not exceed 2]
This picture shows the command I used to run tblastn with the protein sequence to the nucleic acid database.
Finally, attach the
link to the reference article :
https://blog.csdn.net/zxpuls123/article/details/81407277
https://blog .csdn.net/qq_43337286/article/details/103120003