Tutorial de análise de dados de sequenciamento do transcriptoma unicelular Seurat (2) - python (scanpy)
O artigo refere-se ao site oficial escasso para uma explicação mais detalhada.
Este tutorial explora as possibilidades de visualização do scanpy e está dividido em três partes:
Gráfico de dispersão incorporado (por exemplo, UMAP, t-SNE)
Identificação de clusters usando genes marcadores conhecidos
Visualização de genes expressos diferencialmente
Neste tutorial usaremos um conjunto de dados de 10x contendo 68k células de PBMC. Scanpy inclui em sua distribuição uma amostra reduzida deste conjunto de dados, que contém apenas 700 células e 765 genes altamente variáveis. O conjunto de dados foi pré-processado e UMAP calculado.
Neste tutorial também usaremos os seguintes marcadores de literatura:
Células B: CD79A, MS4A1
Plasma: IGJ (JCHAIN)
Células T: CD3D
NK: GNLY, NKG7
Mielóide: CST3, LYZ
Monócitos: FCGR3A
Dendríticos: FCER1A
Gráfico de dispersão incorporado
import scanpy as sc
import pandas as pd
from matplotlib.pyplot import rc_context
sc.set_figure_params(dpi=100, color_map = 'viridis_r')
sc.settings.verbosity = 1
sc.logging.print_header()
Carregar conjunto de dados pbmc
pbmc = sc.datasets.pbmc68k_reduced()
# inspect pbmc contents
pbmc
Visualização da expressão genética e outras variáveis
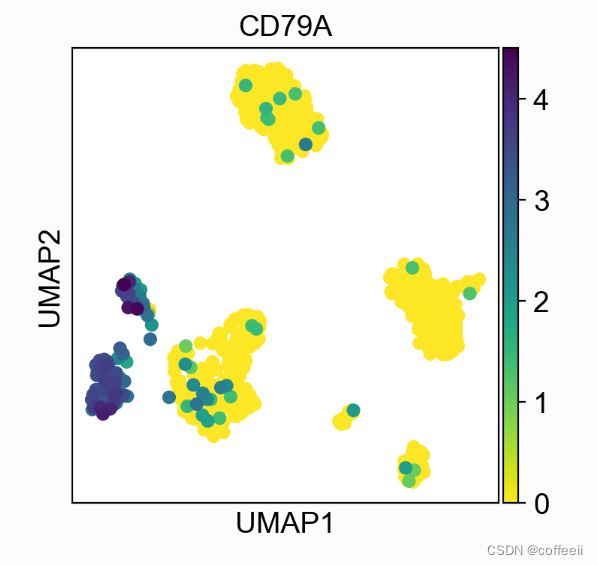
Para gráficos de dispersão, o valor a ser plotado é fornecido como cor do argumento. Pode ser qualquer gene ou qualquer coluna em .obs onde .obs é um DataFrame contendo anotações para cada observação/célula, consulte AnnData para obter mais informações.
# rc_context is used for the figure size, in this case 4x4
with rc_context({'figure.figsize': (4, 4)}):
sc.pl.umap(pbmc, color='CD79A')
Vários valores de cor podem ser fornecidos. No exemplo abaixo, representaremos 6 genes: 'CD79A', 'MS4A1', 'IGJ', CD3D', 'FCER1A' e 'FCGR3A' para entender onde esses genes marcadores são expressos.
Além disso, plotaremos dois outros valores: n_counts que é a contagem UMI para cada célula (armazenada em .obs) e bulk_labels que é um valor categórico contendo os rótulos brutos das células de 10X.
O número de parcelas por linha é controlado pelo parâmetro ncols. O valor máximo desenhado pode ser ajustado usando vmax (da mesma forma, vmin pode ser usado para o valor mínimo). Neste caso, usamos p99, o que significa usar o percentil 99 como valor máximo. Se vmax quiser ser definido separadamente para múltiplas parcelas, o valor máximo pode ser um número ou uma lista de números.
Além disso, estamos usando frameon=False para remover o quadro ao redor do gráfico e s=50 para definir o tamanho do ponto.
with rc_context({'figure.figsize': (3, 3)}):
sc.pl.umap(pbmc, color=['CD79A', 'MS4A1', 'IGJ', 'CD3D', 'FCER1A', 'FCGR3A', 'n_counts', 'bulk_labels'], s=50, frameon=False, ncols=4, vmax='p99')
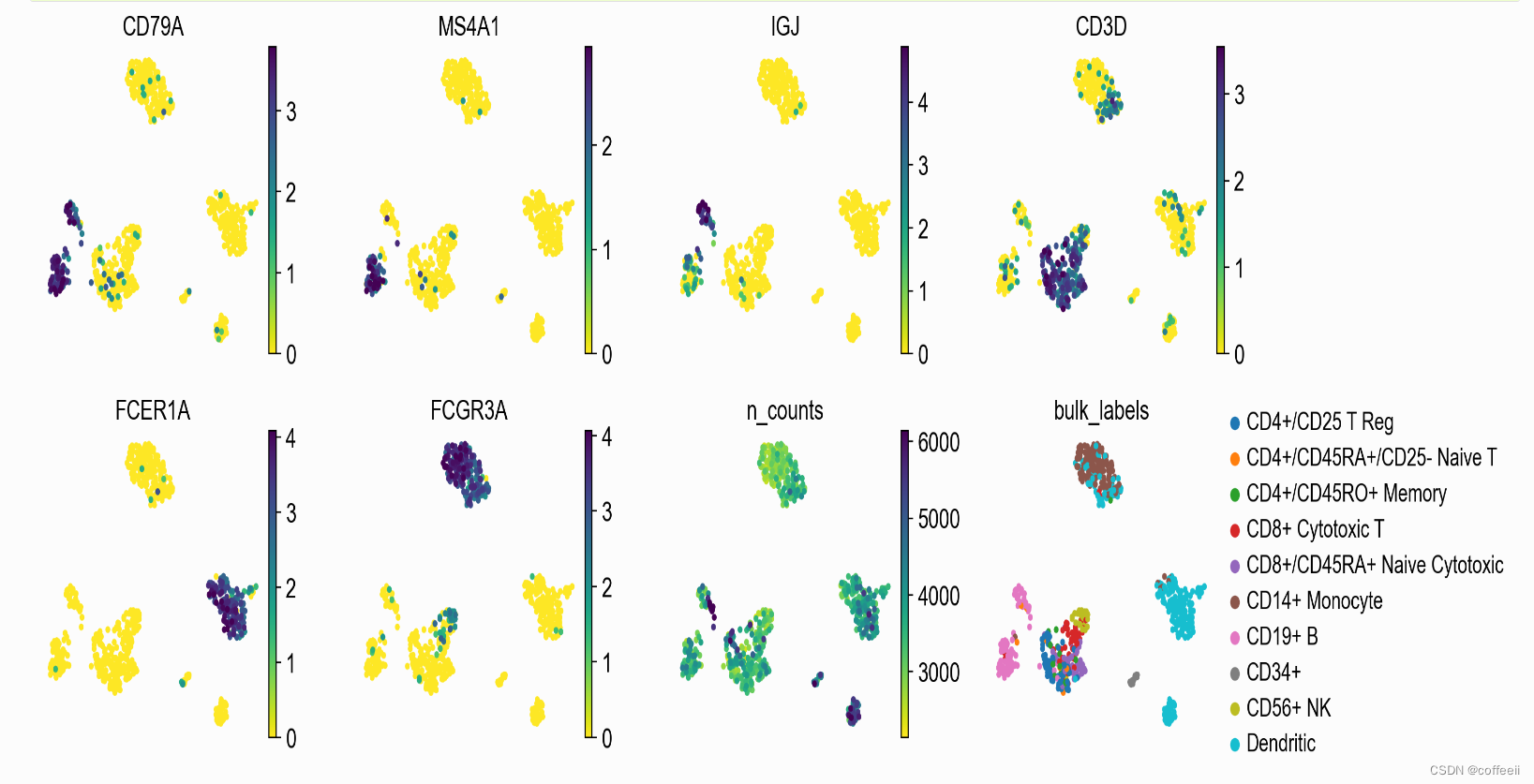
Nesta figura podemos ver o grupo de células que expressam o gene marcador e a concordância com o rótulo celular original.
A função scatterplot tem muitas opções para ajustar a imagem. Por exemplo, podemos visualizar clusters da seguinte forma:
# compute clusters using the leiden method and store the results with the name `clusters`
sc.tl.leiden(pbmc, key_added='clusters', resolution=0.5)
with rc_context({'figure.figsize': (5, 5)}):
sc.pl.umap(pbmc, color='clusters', add_outline=True, legend_loc='on data',
legend_fontsize=12, legend_fontoutline=2,frameon=False,
title='clustering of cells', palette='Set1')

Identificação de cluster baseada em genes marcadores conhecidos
Normalmente, genes marcadores bem conhecidos são necessários para rotular clusters. Usando um gráfico de dispersão, podemos ver a expressão de um gene e potencialmente associá-lo a um cluster. Aqui demonstramos métodos visuais adicionais para associar genes marcadores a clusters usando gráficos de pontos, gráficos de violino, mapas de calor e o que chamamos de “gráficos de trajetória”. Todas essas visualizações resumem as mesmas informações, expressas por clusters, cabendo ao pesquisador a escolha do melhor resultado.
Primeiro, construímos um dicionário com genes marcadores, pois isso permitirá que o scanpy rotule automaticamente os genomas:
marker_genes_dict = {
'B-cell': ['CD79A', 'MS4A1'],
'Dendritic': ['FCER1A', 'CST3'],
'Monocytes': ['FCGR3A'],
'NK': ['GNLY', 'NKG7'],
'Other': ['IGLL1'],
'Plasma': ['IGJ'],
'T-cell': ['CD3D'],
}
Gráfico de pontos
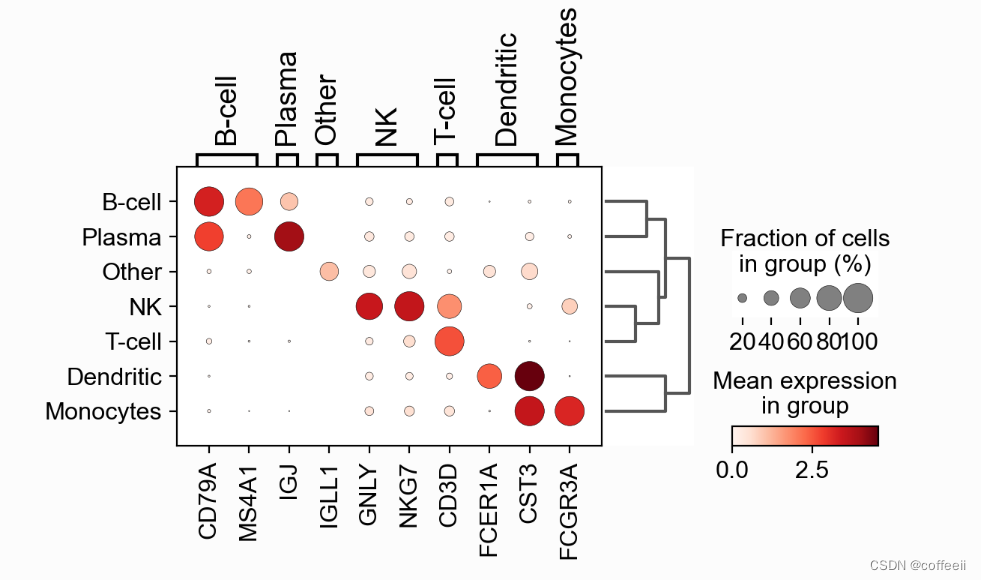
Uma maneira rápida de examinar a expressão desses genes em cada cluster é usar um gráfico de pontos. Esse tipo de gráfico resume dois tipos de informação: a cor representa a expressão média em cada categoria (neste caso, cada cluster) e o tamanho do ponto representa a proporção de células na categoria que expressam o gene.
Além disso, é útil adicionar um dendograma ao gráfico para agrupar clusters semelhantes. O clustering hierárquico é calculado automaticamente usando a correlação de componentes do PCA entre clusters.
sc.pl.dotplot(pbmc, marker_genes_dict, 'clusters', dendrogram=True)
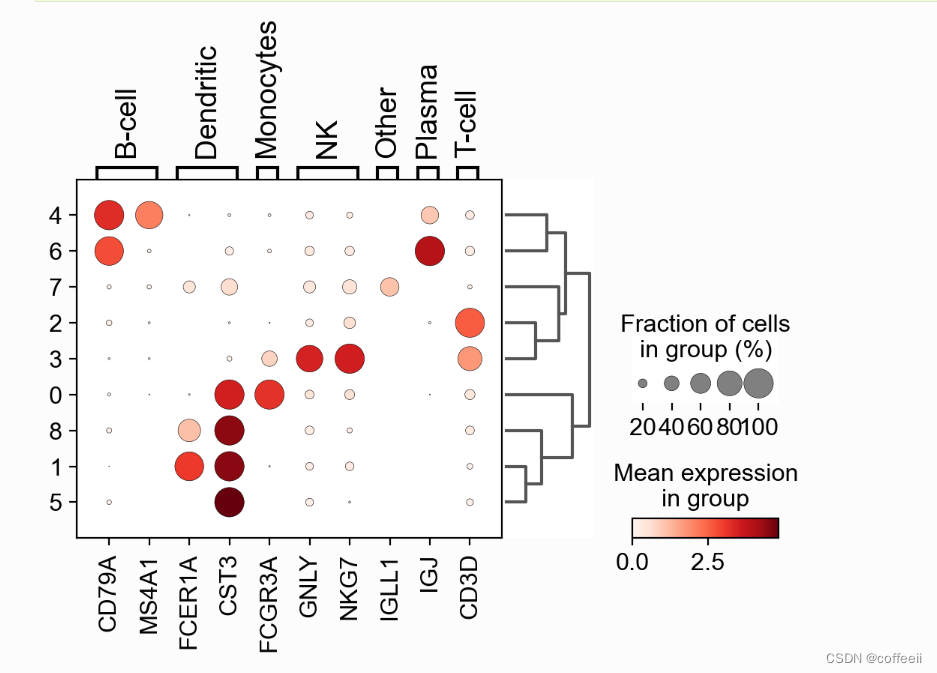
Usando este diagrama, podemos ver que o cluster 4 corresponde às células B, o cluster 2 corresponde às células T e assim por diante. Essas informações podem ser usadas para anotar células manualmente da seguinte maneira:
# create a dictionary to map cluster to annotation label
cluster2annotation = {
'0': 'Monocytes',
'1': 'Dendritic',
'2': 'T-cell',
'3': 'NK',
'4': 'B-cell',
'5': 'Dendritic',
'6': 'Plasma',
'7': 'Other',
'8': 'Dendritic',
}
# add a new `.obs` column called `cell type` by mapping clusters to annotation using pandas `map` function
pbmc.obs['cell type'] = pbmc.obs['clusters'].map(cluster2annotation).astype('category')
sc.pl.dotplot(pbmc, marker_genes_dict, 'cell type', dendrogram=True)
sc.pl.umap(pbmc, color='cell type', legend_loc='on data',
frameon=False, legend_fontsize=10, legend_fontoutline=2)
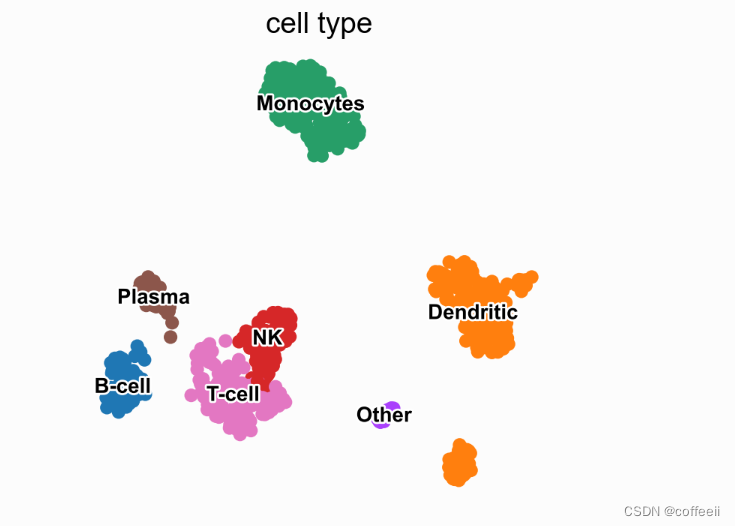
Diagrama de violino
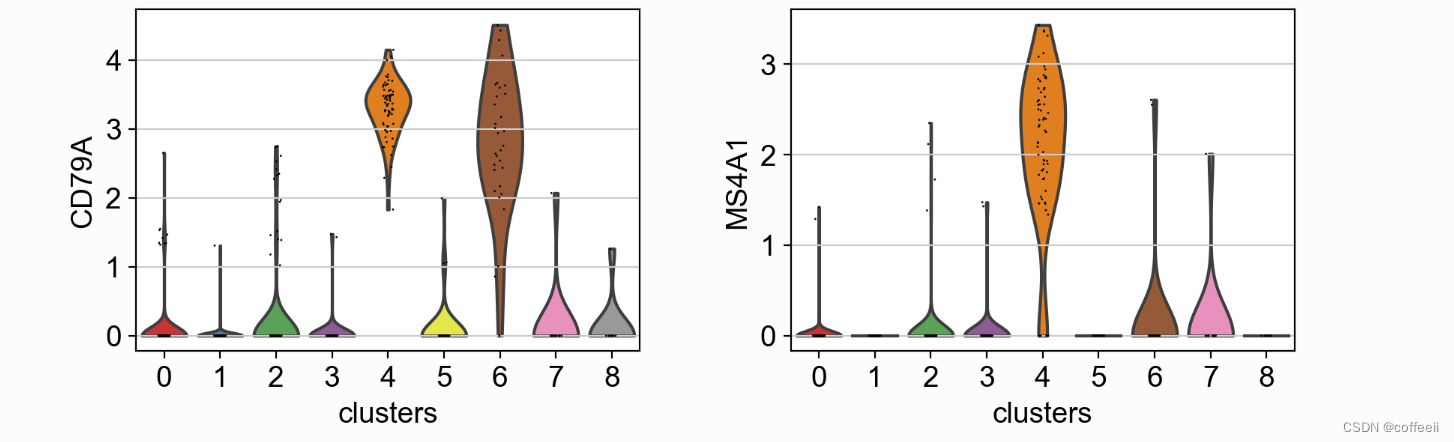
Outra forma de explorar marcadores é usar plotagens de violino. Aqui, podemos ver a expressão de CD79A nos clusters 5 e 8 e MS4A1 no cluster 5. Comparados aos gráficos de pontos, os gráficos de violino nos dão uma ideia da distribuição dos valores de expressão gênica entre as células.
with rc_context({'figure.figsize': (4.5, 3)}):
sc.pl.violin(pbmc, ['CD79A', 'MS4A1'], groupby='clusters' )
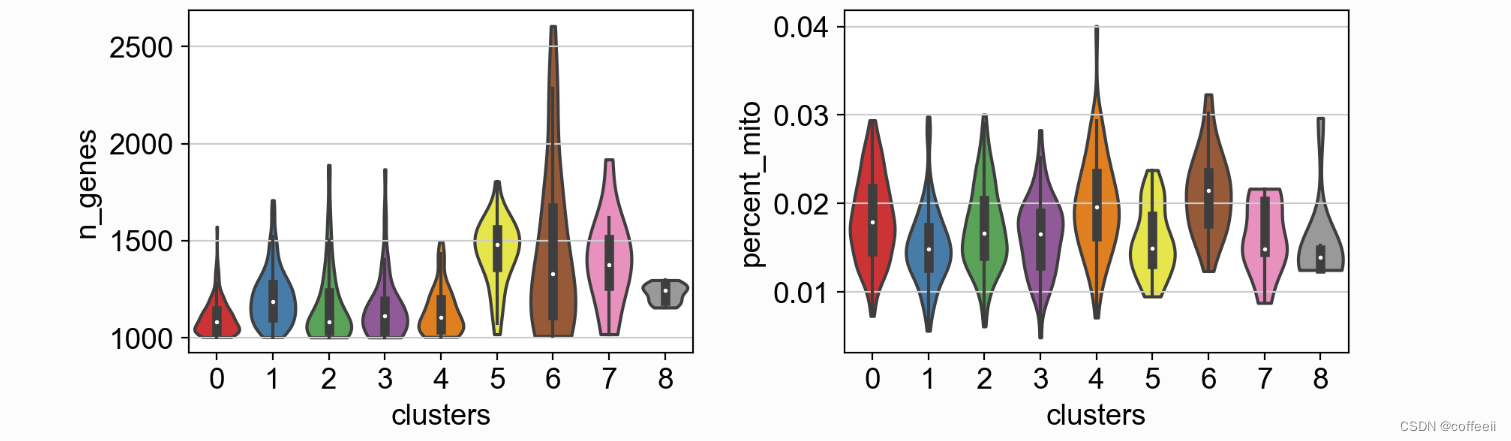
with rc_context({'figure.figsize': (4.5, 3)}):
sc.pl.violin(pbmc, ['n_genes', 'percent_mito'], groupby='clusters', stripplot=False, inner='box') # use stripplot=False to remove the internal dots, inner='box' adds a boxplot inside violins
enredo de violino empilhado
Veja também o gráfico do violino sc.pl.stacked_violin para todos os genes marcadores que usamos. Como antes, um dendograma foi adicionado para agrupar clusters semelhantes
ax = sc.pl.stacked_violin(pbmc, marker_genes_dict, groupby='clusters', swap_axes=False, dendrogram=True)
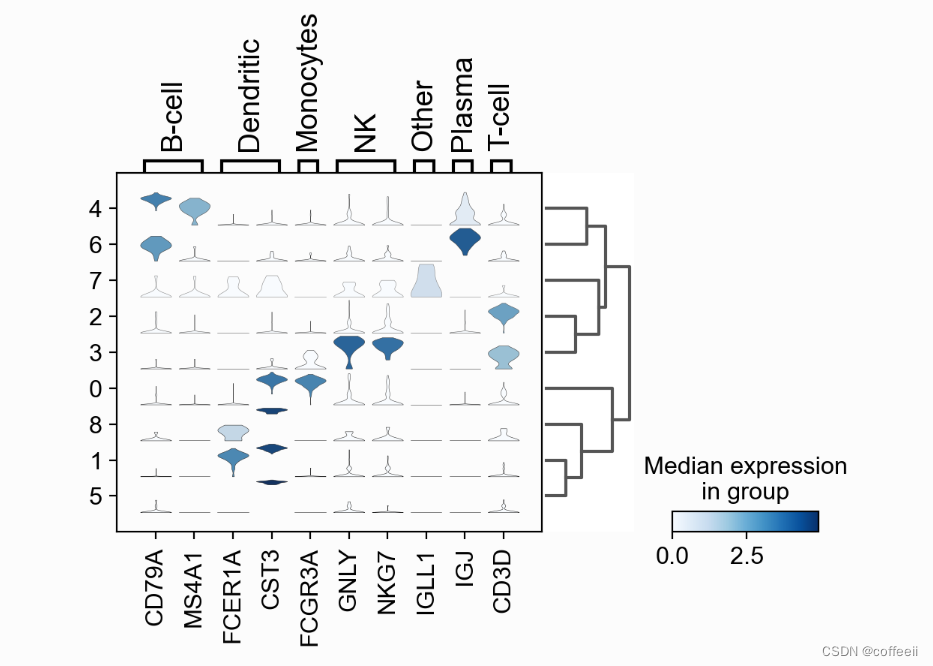
diagrama de matriz
Uma maneira fácil de visualizar a expressão gênica é usar .Este é um mapa de calor do valor médio da expressão para cada gene agrupado por categoria. Esse tipo de gráfico mostra basicamente as mesmas informações que as cores em um gráfico de pontos. gráfico de matriz
Aqui, a expressão do gene é dimensionada de 0 a 1, que é a expressão média máxima e 0 a mínima.
sc.pl.matrixplot(pbmc, marker_genes_dict, 'clusters', dendrogram=True, cmap='Blues', standard_scale='var', colorbar_title='column scaled\nexpression')
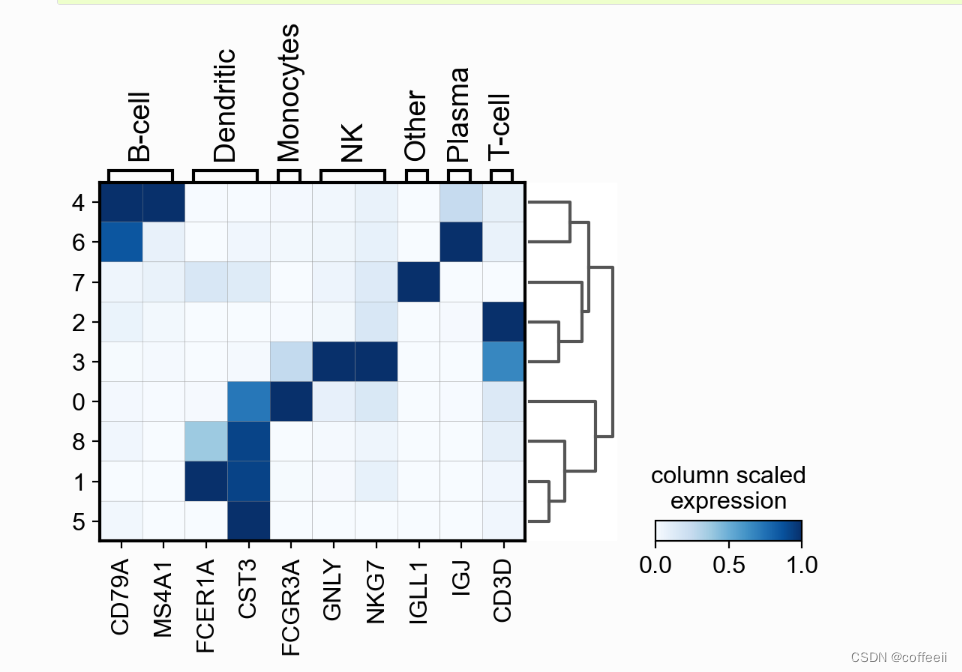
Outra opção útil é usar sc.pp.scale. Aqui armazenamos essas informações na camada de escala. Depois disso ajustamos os valores mínimo e máximo do gráfico e usamos um mapa de cores divergente (neste caso RdBu_r significa_r é o oposto).
# scale and store results in layer
pbmc.layers['scaled'] = sc.pp.scale(pbmc, copy=True).X
sc.pl.matrixplot(pbmc, marker_genes_dict, 'clusters', dendrogram=True,
colorbar_title='mean z-score', layer='scaled', vmin=-2, vmax=2, cmap='RdBu_r')
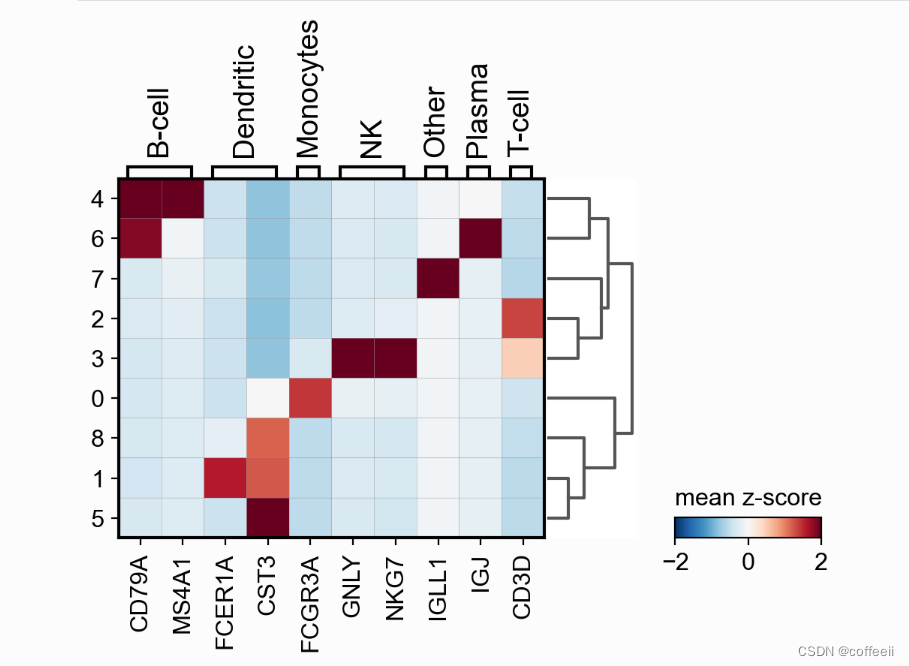
mapa de calor
O mapa de calor não colapsa as células como no gráfico anterior. Em vez disso, cada célula é exibida em uma linha (ou coluna se swap_axes=True ). Você pode adicionar informações de agrupamento e exibir sc.pl.umap usando o mesmo código de cores encontrado ou qualquer outra incorporação.
ax = sc.pl.heatmap(pbmc, marker_genes_dict, groupby='clusters', cmap='viridis', dendrogram=True)
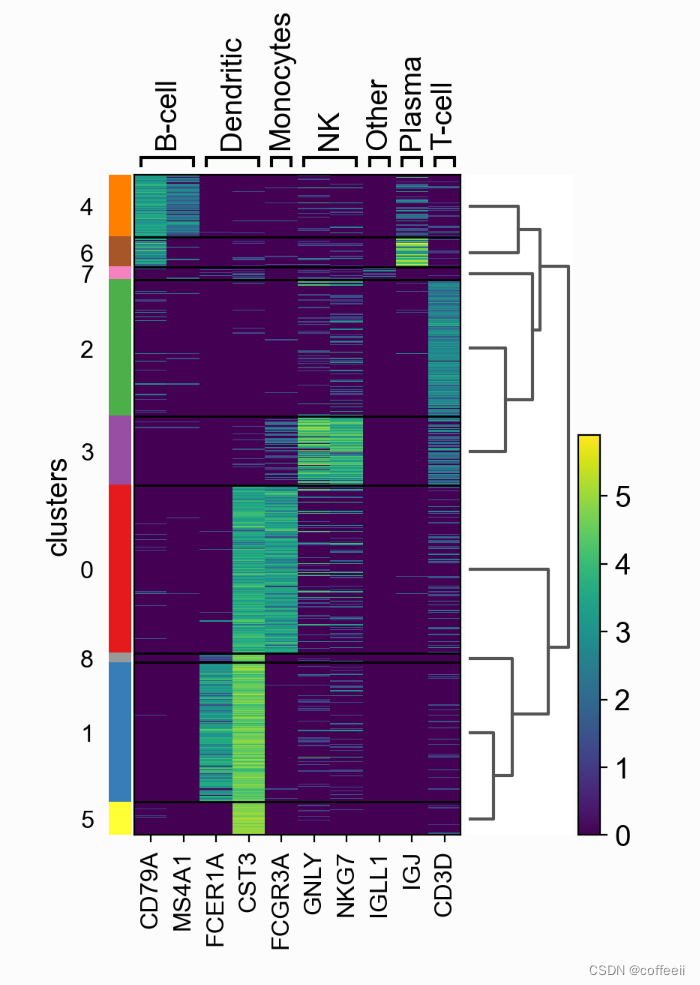
Os mapas de calor também podem ser plotados em dados dimensionados. Na próxima imagem, semelhante à imagem da matriz anterior, os valores mínimo e máximo são ajustados e um mapa de cores divergente é usado.
ax = sc.pl.heatmap(pbmc, marker_genes_dict, groupby='clusters', layer='scaled', vmin=-2, vmax=2, cmap='RdBu_r', dendrogram=True, swap_axes=True, figsize=(11,4))
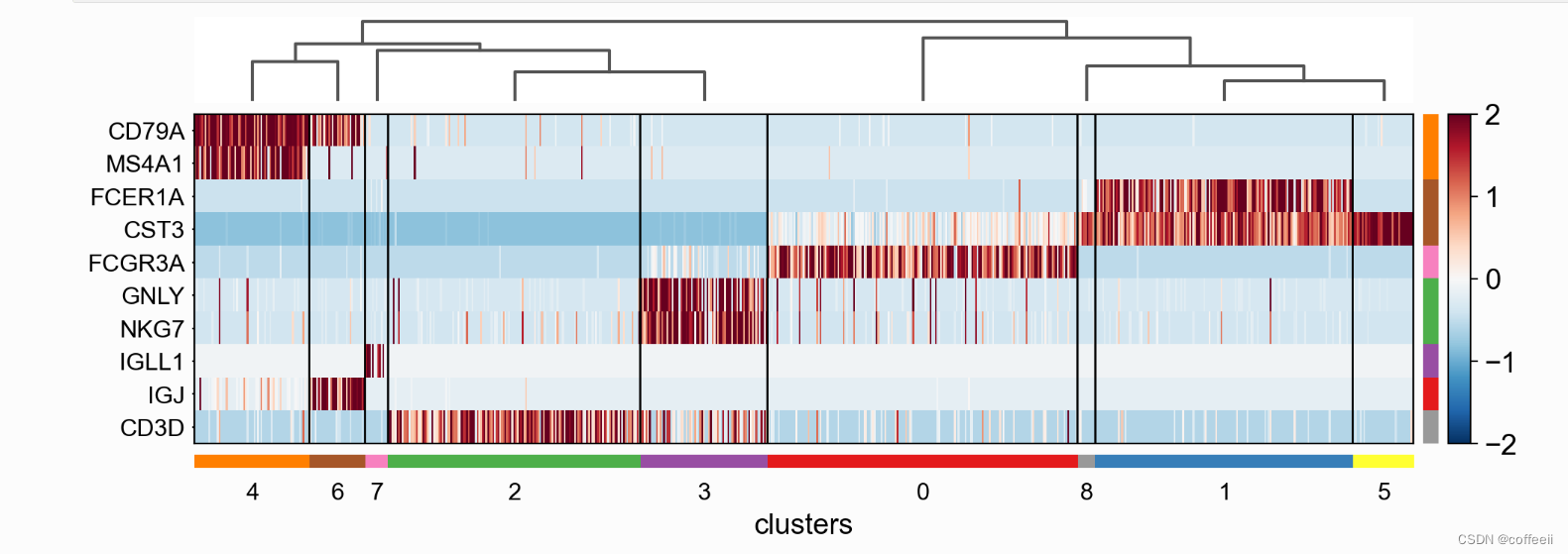
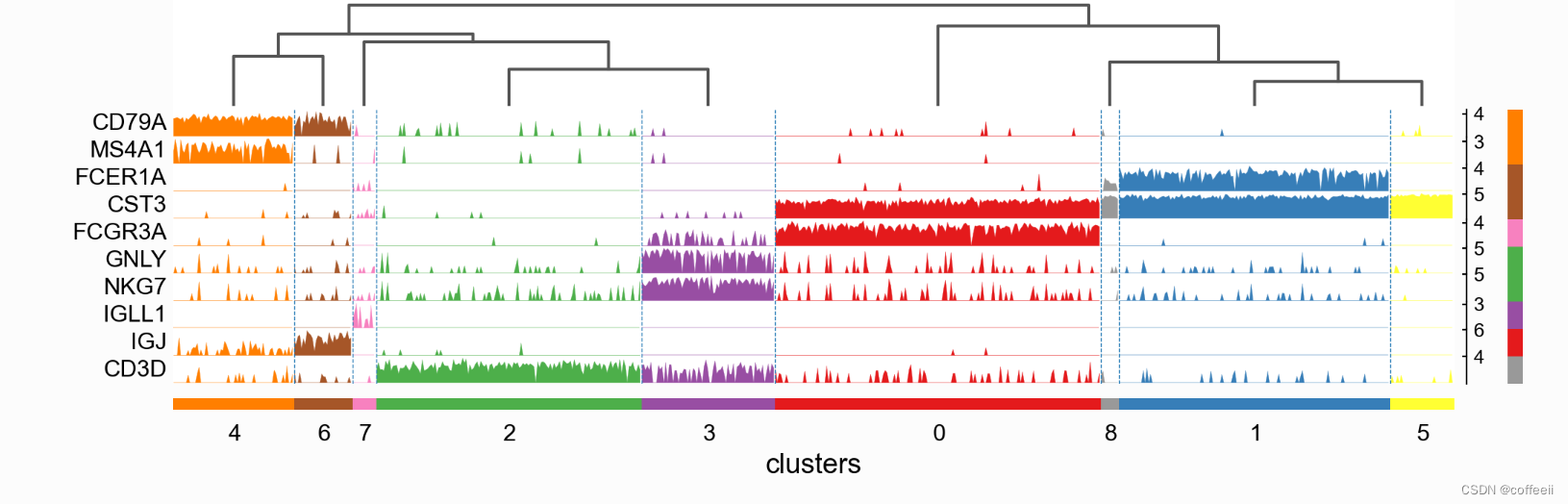
Diagrama de trajetória
Os gráficos de trajetória exibem as mesmas informações que os mapas de calor, mas a expressão genética é representada pela altura em vez de uma escala de cores.
ax = sc.pl.tracksplot(pbmc, marker_genes_dict, groupby='clusters', dendrogram=True)
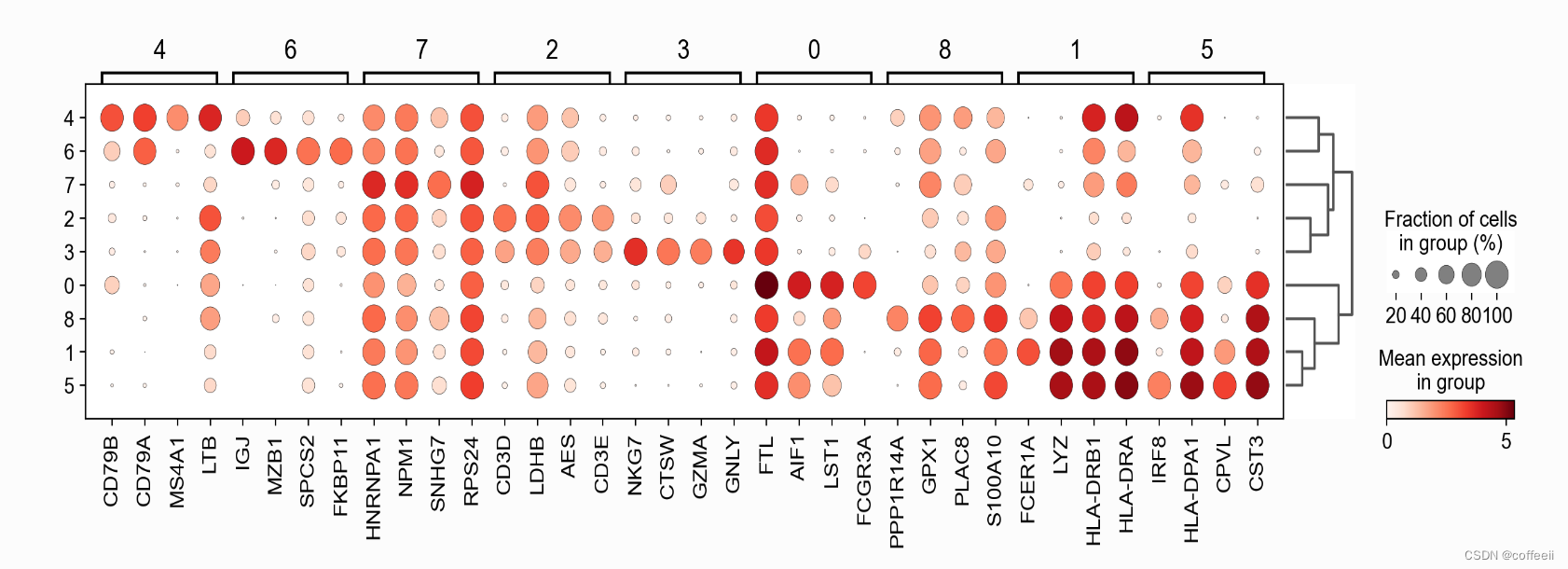
Visualização de genes marcadores
Em vez de caracterizar clusters por assinaturas genéticas conhecidas, como feito anteriormente, podemos identificar genes que são expressos diferencialmente em clusters ou grupos.
Para identificar genes expressos diferencialmente, executamos sc.tl.rank_genes_groups . A função pegará cada grupo de células e comparará a distribuição de cada gene em um grupo com a distribuição em todas as outras células que não estão nesse grupo. Aqui, usaremos os rótulos celulares originais fornecidos por 10x para identificar genes marcadores para esses tipos de células.
sc.tl.rank_genes_groups(pbmc, groupby='clusters', method='wilcoxon')
Visualizando genes marcadores usando gráficos de pontos
A visualização do gráfico de pontos ajuda a compreender o perfil dos genes que apresentam expressão diferencial. Para tornar a imagem resultante mais compacta, usaremos n_genes=4 para exibir apenas os 4 principais genes de pontuação.
sc.pl.rank_genes_groups_dotplot(pbmc, n_genes=4)
Para uma melhor representação, podemos representar graficamente a mudança logarítmica em vez da expressão gênica. Além disso, desejamos nos concentrar em genes com uma alteração logarítmica> = 3 entre a expressão do tipo de célula e o restante das células.
Neste caso, definimos valores_to_plot='logfoldchanges' e min_logfoldchange=3.
Como a mudança log-fold é uma escala divergente, também ajustamos os valores mínimo e máximo a serem plotados e usamos um mapa de cores divergente. Observe que é difícil distinguir as populações de células T na imagem abaixo.
sc.pl.rank_genes_groups_dotplot(pbmc, n_genes=4, values_to_plot='logfoldchanges', min_logfoldchange=3, vmax=7, vmin=-7, cmap='bwr')
concentre-se em grupos específicos
A seguir, usamos um gráfico de pontos com foco em apenas dois grupos (a opção de grupo também está disponível para gráficos de violino, mapas de calor e gráficos de matriz). Aqui configuramos para que neste caso ele exiba todos os genes com n_genes=30 no máximo 30. min_logfoldchange=4
sc.pl.rank_genes_groups_dotplot(pbmc, n_genes=30, values_to_plot='logfoldchanges', min_logfoldchange=4, vmax=7, vmin=-7, cmap='bwr', groups=['1', '5'])
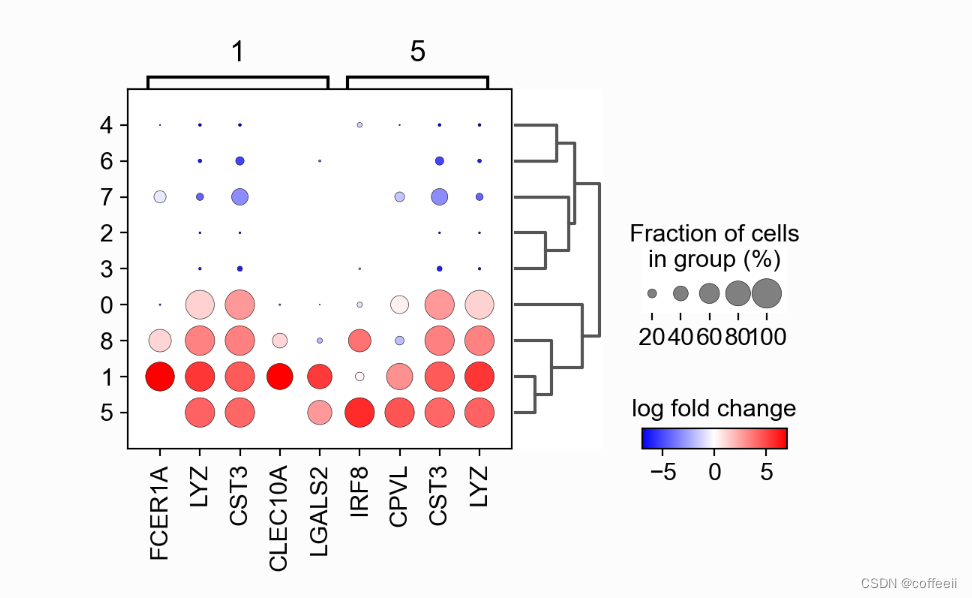
Use o Matrixplot para visualizar genes marcadores
Para a imagem abaixo, utilizamos o valor de "escala" calculado anteriormente (armazenado na camada dimensionada) e utilizamos um mapa de cores divergente. \
sc.pl.rank_genes_groups_matrixplot(pbmc, n_genes=3, use_raw=False, vmin=-3, vmax=3, cmap='bwr', layer='scaled')
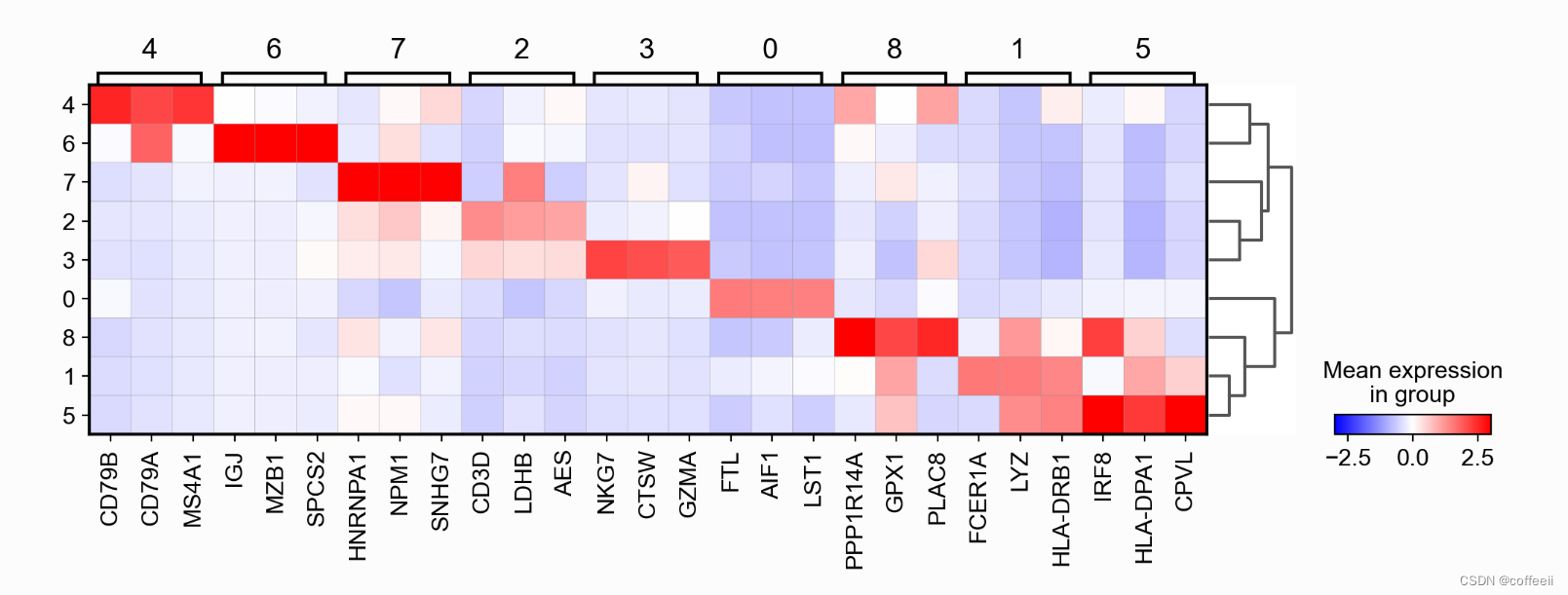
Visualizando genes marcadores usando gráficos de violino empilhados
sc.pl.rank_genes_groups_stacked_violin(pbmc, n_genes=3, cmap='viridis_r')
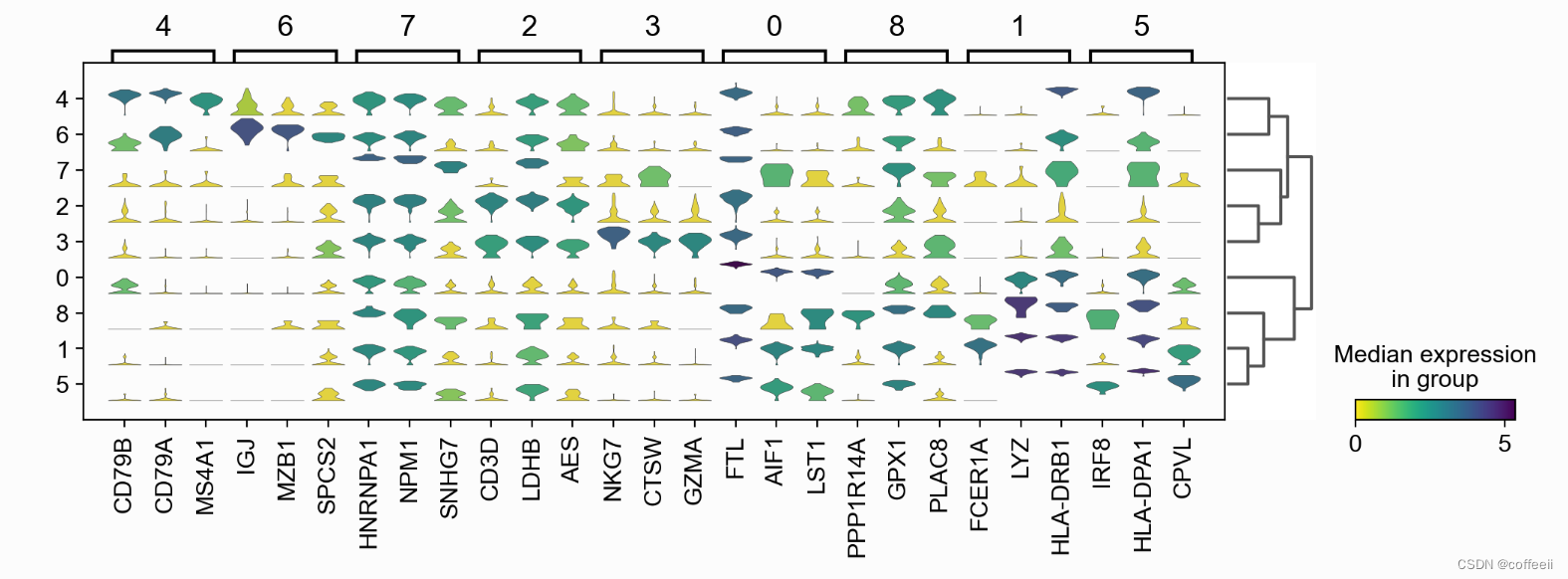
Visualizando genes marcadores usando mapas de calor
sc.pl.rank_genes_groups_heatmap(pbmc, n_genes=3, use_raw=False, swap_axes=True, vmin=-3, vmax=3, cmap='bwr', layer='scaled', figsize=(10,7), show=False);
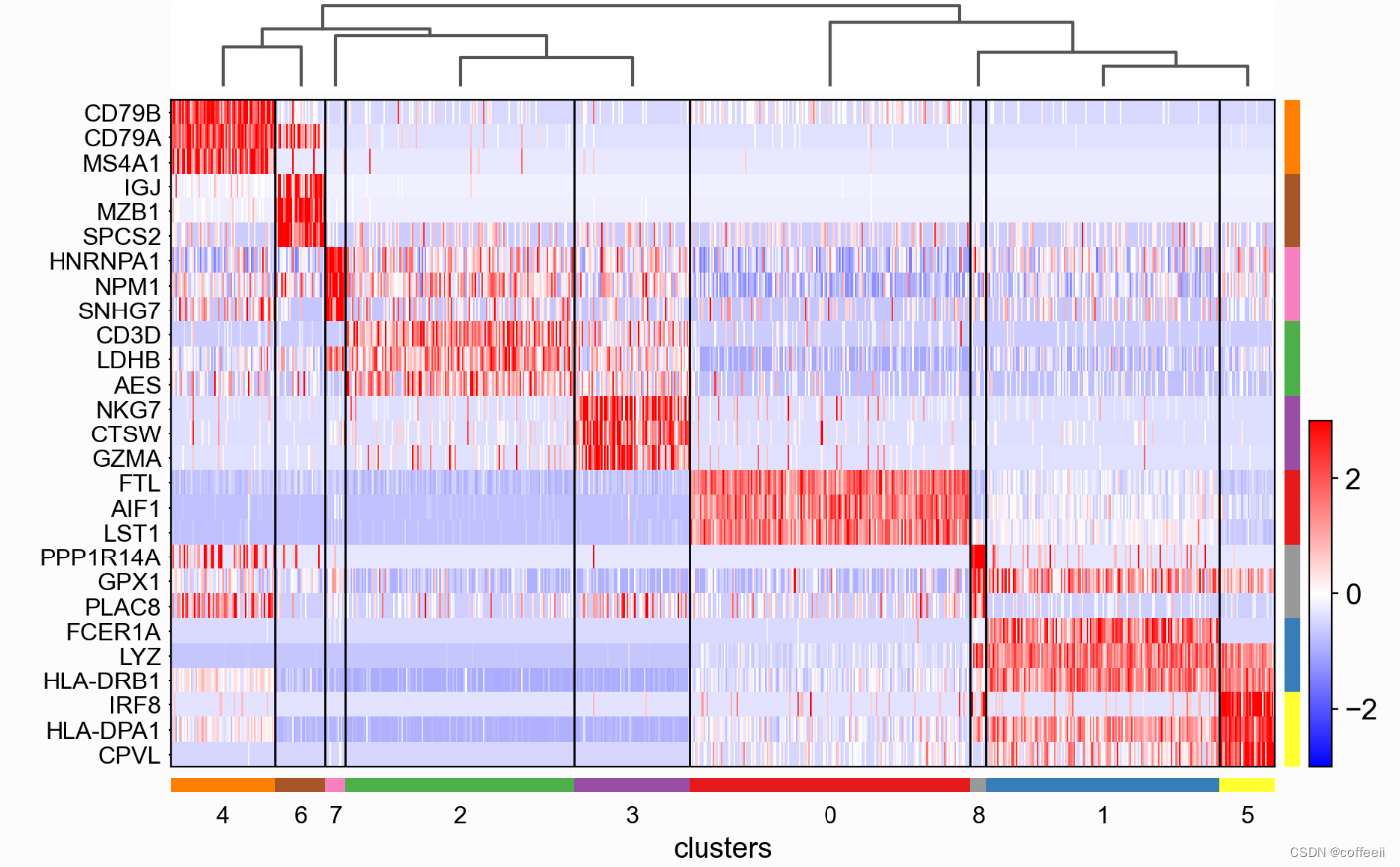
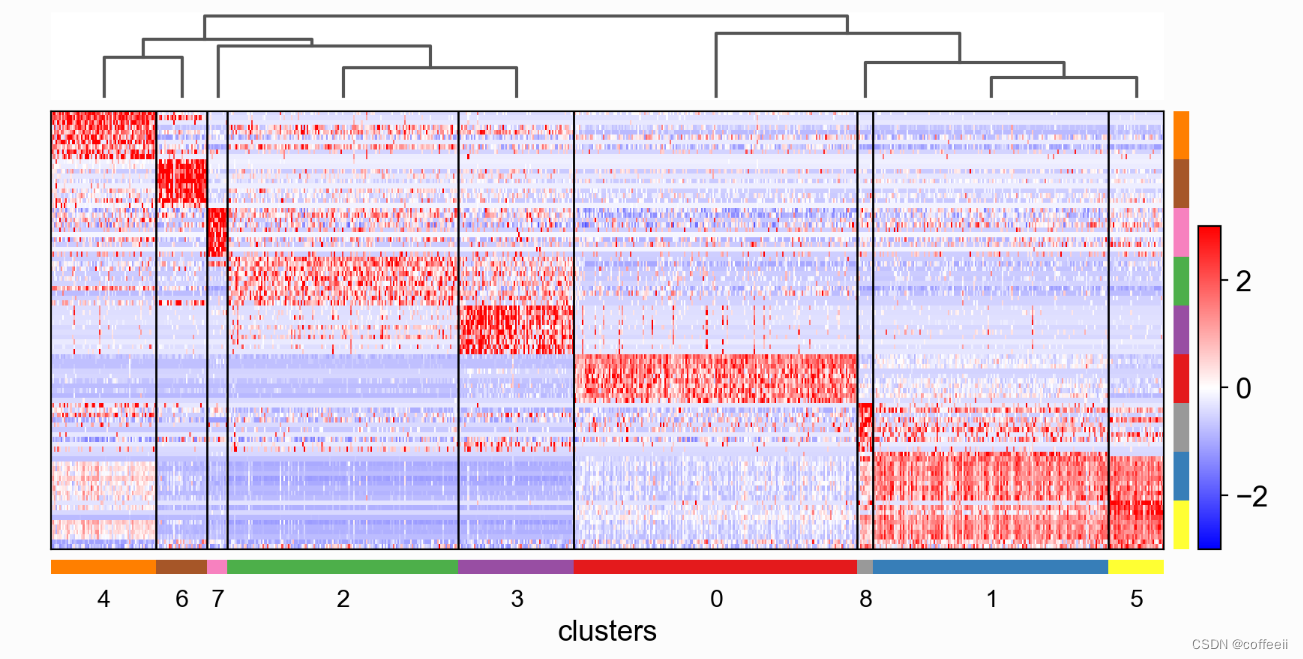
10 genes são mostrados por categoria, com rótulos de genes desativados e eixos trocados. Observe que ao trocar imagens, os códigos de cores das categorias aparecem em vez de “colchetes”.
sc.pl.rank_genes_groups_heatmap(pbmc, n_genes=10, use_raw=False, swap_axes=True, show_gene_labels=False,
vmin=-3, vmax=3, cmap='bwr')
Visualizando genes marcadores usando gráficos de trajetória
sc.pl.rank_genes_groups_tracksplot(pbmc, n_genes=3)
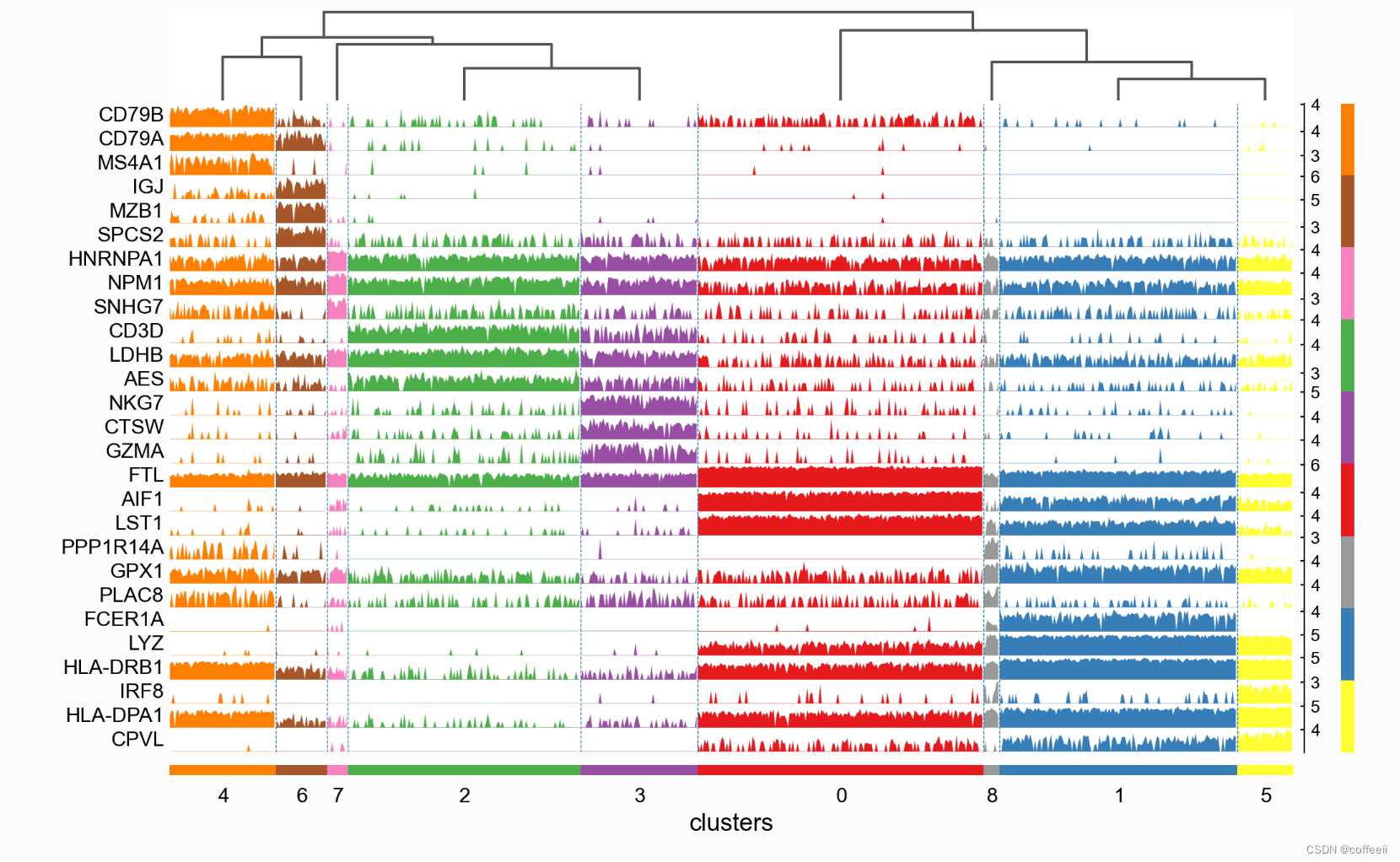
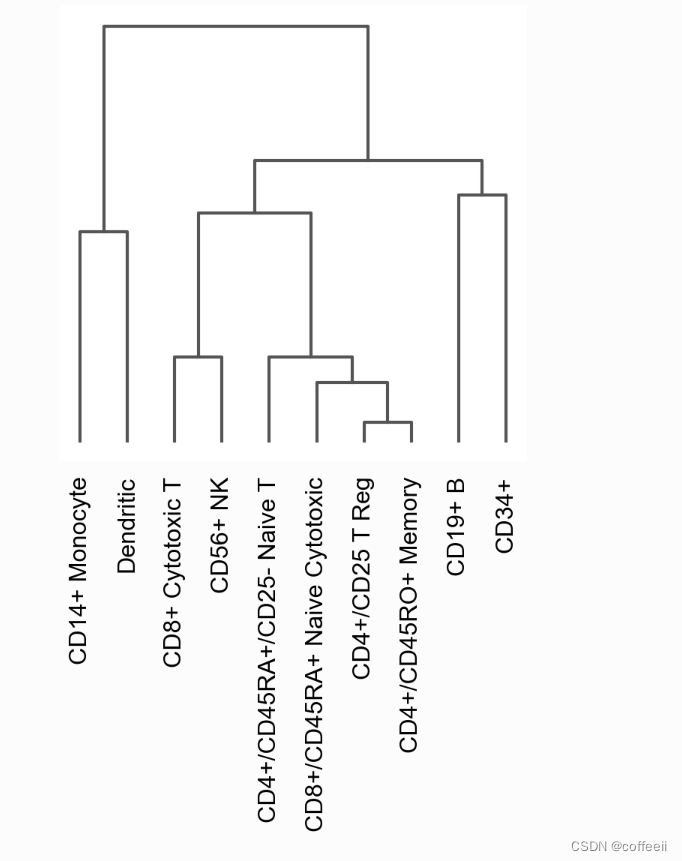
Opções de dendograma
A maioria das visualizações pode usar um diagrama de árvore para organizar categorias. No entanto, o dendograma também pode ser desenhado de forma independente da seguinte forma:
# compute hierarchical clustering using PCs (several distance metrics and linkage methods are available).
sc.tl.dendrogram(pbmc, 'bulk_labels')
ax = sc.pl.dendrogram(pbmc, 'bulk_labels')