Die DNA-Cytosin-Methylierung (5 mC) ist eine wichtige epigenetische Modifikation, die an wichtigen Prozessen wie der zellspezifischen Regulierung der Genexpression und der Aufrechterhaltung der Genomstabilität beteiligt ist. In Säugetierzellen kann die DNA-Cytosin-Methylierung durch Proteine der TET-Familie oxidiert und anschließend demethyliert werden, wodurch die Cytosin-Modifikation im Genom dynamisch reguliert wird (Abbildung 1a, b)[1] . Die Cytosin-Hydroxymethylierungsmodifikation (5hmC) ist das am häufigsten vorkommende Demethylierungszwischenprodukt im Genom (insbesondere in Nervenzellen) und kann eine wichtige Funktion spielen [2] . Da die herkömmliche Bisulfit-Sequenzierungstechnologie (Bisulfit-Sequenzierung) jedoch nicht zwischen 5mC- und 5hmC-Modifikationen unterscheiden kann (Abbildung 1c) , entwickelte das Forschungsteam 2018 die ACE-seq-Technologie, um 5hmC-Modifikationen mit Einzelbasenauflösung zu identifizieren [3] . Allerdings wie Der quantitative Nachweis dieser beiden DNA-Modifikationen auf Einzelzellebene ist immer noch eine Herausforderung.
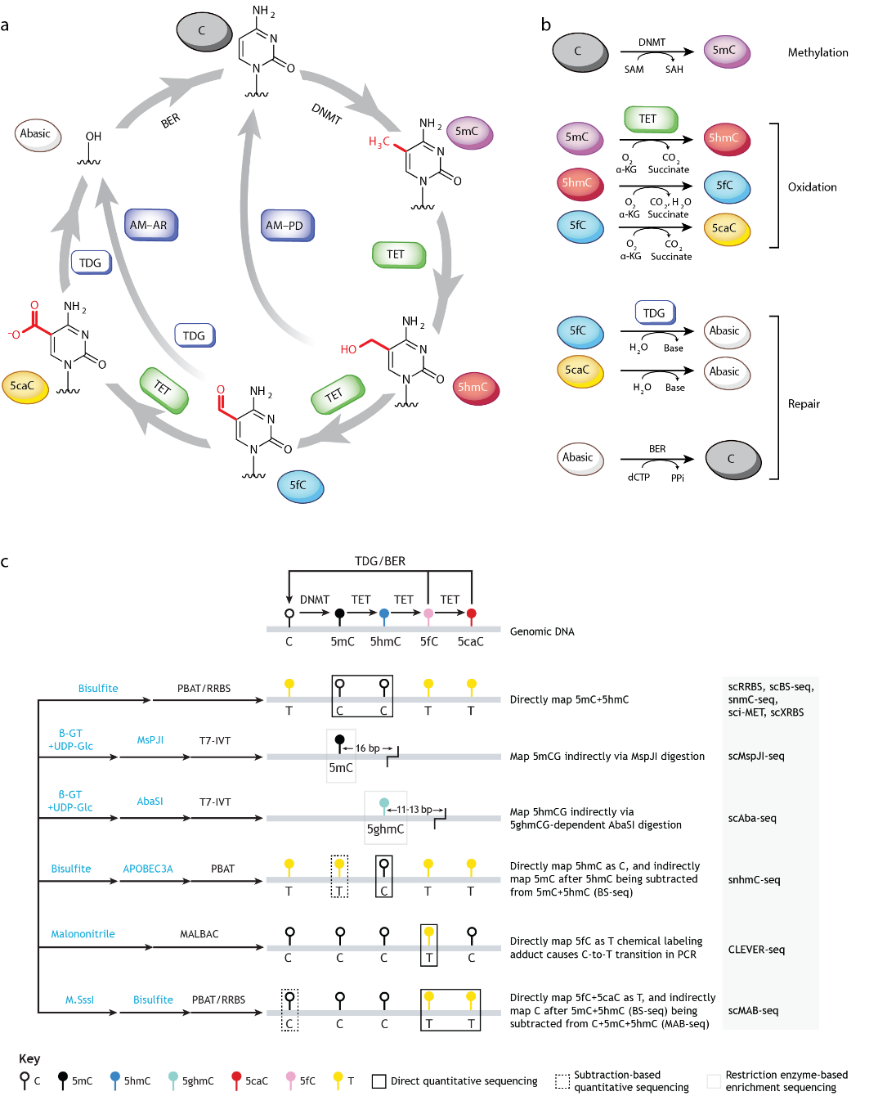
Abb. 1 Dynamische Regulierung der DNA-Cytosin-Methylierung und sequenzierungsbasierte Detektionstechnologie [1,2] .
Am 28. August 2023 veröffentlichte das Labor von Wu Hao an der University of Pennsylvania in der Fachzeitschrift Nature Biotechnology eine Forschungsarbeit mit dem Titel „ Joint-Single-Cell Profiling löst 5mC und 5hmC auf und enthüllt ihre unterschiedlichen genregulierenden Wirkungen“ , in der über einen gleichzeitigen Nachweis von genomischer DNA 5hmC und 5mC berichtet wird Eine modifizierte Einzelzell-Sequenzierungstechnologie, Joint-snhmC-seq (vollständiger Name: Joint Single-Nucleus (Hydroxy) Methylcytosine Sequencing).

Der Kern dieser Technologie ist die Umwandlung von unmodifiziertem Cytosin (C) und methyliertem Cytosin (5mC) in Das Forschungsteam fügte dem Konstruktionsprozess der snmC-seq-Bibliothek zunächst den Schritt der enzymatischen Desaminierung hinzu [4] und entwickelte die snhmC-seq-Technologie zum Nachweis von 5hmC-Stellen in einzelnen Zellen (Abb. 2a) . Durch die systematische Optimierung der Bedingungen der Desaminierungsreaktion und der Schlüsselschritte des Bibliotheksaufbaus verbesserten die Autoren die Nachweisempfindlichkeit und -stabilität (snhmC-seq2-Technologie) und entwickelten die Joint-snhmC-seq-Technologie für den gleichzeitigen Nachweis von 5mC und 5hmC weiter (Abbildung 2b). ) . Mithilfe der Joint-snhmC-seq-Technologie bestimmten die Autoren die Modifikationsniveaus von 5hmC und echtem 5mC in verschiedenen Zelltypen in der Großhirnrinde der Maus, untersuchten weiter die Beziehung zwischen diesen beiden Modifikationen und den Gentranskriptionsniveaus und stellten fest, dass einige Zelltypen Spezifisch exprimierte Long-Gene neigen unerwarteterweise dazu, höhere 5mC-Werte im Genkörper aufrechtzuerhalten und einen aktiven Transkriptionszustand aufrechtzuerhalten.
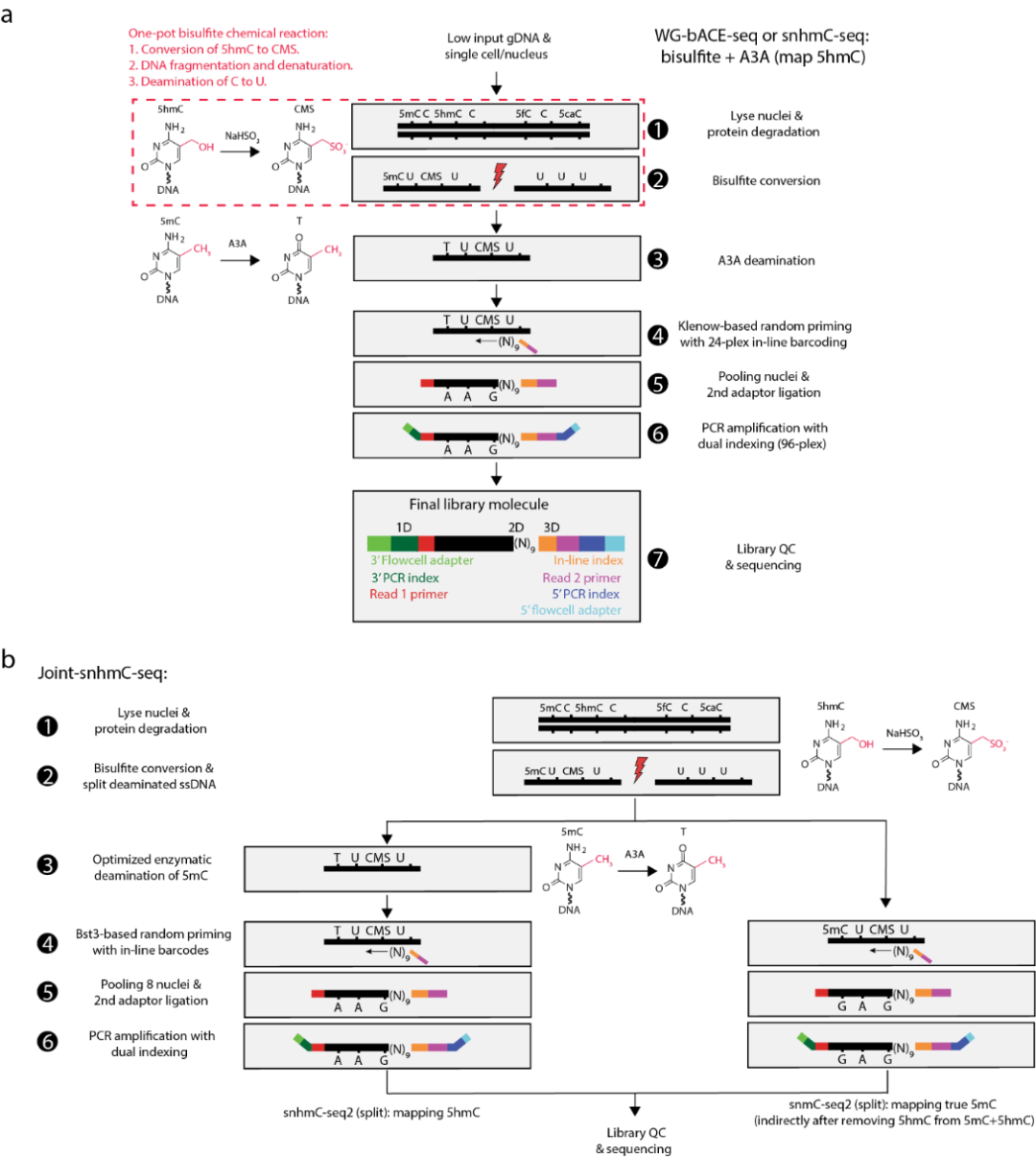
Abbildung 2 Flussdiagramm von snhmC-seq und Joint-snhmC-seq.
Um die Genauigkeit der Zellclusterergebnisse der Joint-snhmC-seq-Technologie zu bewerten, verwendeten die Autoren zunächst die immunfluoreszenzgestützte Kernsortierungstechnik, um die Kerne der Großhirnrinde der Maus in nicht-neurale Zellen (NeuN-) zu unterteilen. Hemmende Neuronen (NeuN+ & Neurod6-) und erregende Neuronen (NeuN+ & Neurod6+) und führen Sie dann eine Joint-snhmC-seq-Analyse an den gesammelten Kernen durch. Mithilfe der Ähnlichkeit der CG-Methylierung einzelner Zellen zur Clusterbildung identifizierten die Autoren 6 Zelltypen, und die Clusterergebnisse stimmten in hohem Maße mit den Ergebnissen der Kernsortierung überein. Darüber hinaus analysierten die Autoren gemeinsam die beiden Module der Joint-snhmC-seq-Daten, um die Konzentrationen von Cytosin (unmodifiziertes C), 5mC und 5hmC in verschiedenen Zelltypen quantitativ zu analysieren. In Übereinstimmung mit früheren Studien waren die 5hmC-Spiegel (23,8–29,4 %) in den drei Arten neuronaler Zellen deutlich höher als die in den drei Arten nicht-neuraler Zellen. Gleichzeitig stellten die Autoren auch ein Phänomen fest, über das nicht berichtet wurde: Es gibt signifikante Unterschiede in den 5hmC-Spiegeln zwischen verschiedenen nicht-neuronalen Zellen [Astrozyten, 12,7 %; Mikroglia, 3,58 %].
Die Autoren gruppierten neuronale Zellen mithilfe der Ähnlichkeit der mononukleären CH-Methylierung neu, identifizierten außerdem acht Zellsubtypen und quantifizierten die 5mC- und 5hmC-Spiegel in diesen Zellsubtypen.
Zusammenfassend ist Joint-snhmC-seq eine neue Technologie, die DNA-Cytosin-5mC- und 5hmC-Modifikationen auf Einzelzellebene genau quantifizieren kann. Im Vergleich zur bestehenden 5hmC-Sequenzierungstechnologie [5,6] bietet diese neue Technologie die Vorteile einer geringen Probeneingabe, einer hohen Genauigkeit und der gleichzeitigen Erkennung von zwei Modifikationen. Es wird erwartet, dass diese Technologie zur Untersuchung wichtiger biologischer Fragen wie der Rolle der 5mC- und 5hmC-Modifikation bei der Entwicklung und Reifung des Gehirns sowie bei Prozessen im Zusammenhang mit der Krankheitsentstehung eingesetzt wird.
Professor Wu Hao von der University of Pennsylvania ist der korrespondierende Autor und Dr. Emily Fabyanic (Absolventin), Dr. Hu Peng (derzeit Professor an der School of Fisheries and Life Sciences der Shanghai Ocean University) und Dr. Qiu Qi Co-Erstautoren. Dr. Emily Fabyanic und Dr. Qiu Qi haben die Methode zum Aufbau der Joint-snhmC-seq-Bibliothek etabliert und optimiert, und Dr. Hu Peng hat den Datenanalyseprozess Joint-snhmC-seq entwickelt. Auch die Labore von Zhaolan Zhou und Rahul Kohli an der University of Pennsylvania leisteten wichtige Beiträge zur Forschung.
Ursprünglicher Link:
https://www.nature.com/articles/s41587-023-01909-2
Hersteller: Elf
Verweise
1. Kohli, R. & Zhang, Y. TET-Enzyme, TDG und die Dynamik der DNA-Demethylierung. Natur 502, 472–479 (2013).
2. Wei, A. & Wu, H. DNA-Methylomdynamik von Säugetieren: Mechanismen, Funktionen und neue Grenzen. Entwicklung 149, dev182683 (2022).
3. Schutsky, EK et al. Zerstörungsfreie, basenaufgelöste Sequenzierung von 5-Hydroxymethylcytosin mithilfe einer DNA-Deaminase. Nat Biotechnol 36, 1083-1090 (2018).
4. Luo, C. et al. Einzelzell-Methylome identifizieren neuronale Subtypen und regulatorische Elemente im Cortex von Säugetieren. Wissenschaft 357, 600–604 (2017).
5. Yu, M. et al. Basenauflösende Analyse von 5-Hydroxymethylcytosin im Säugetiergenom. Zelle 149, 1368–1380 (2012).
6. Booth, MJ et al. Quantitative Sequenzierung von 5-Methylcytosin und 5-Hydroxymethylcytosin mit Einzelbasenauflösung. Wissenschaft 336, 934–937 (2012).
Frühere Produkte (klicken Sie auf das Bild, um direkt zum Text des entsprechenden Tutorials zu gelangen)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|