Version diapositive-séquence
base de données
Nous analysons ici un ensemble de données généré à l’aide de Slide-seq v2 de l’hippocampe de souris. Ce didacticiel suivra en grande partie la même structure que la vignette spatiale de 10x Visium Data, mais est conçu pour fournir une démonstration spécifique aux données Slide-seq.
Vous pouvez utiliser notre package SeuratData pour accéder facilement aux données comme indiqué ci-dessous. Une fois l'ensemble de données installé, vous pouvez taper ?ssHippo pour voir les commandes utilisées pour créer des objets Seurat.
InstallData("ssHippo")
slide.seq <- LoadData("ssHippo")
prétraitement des données
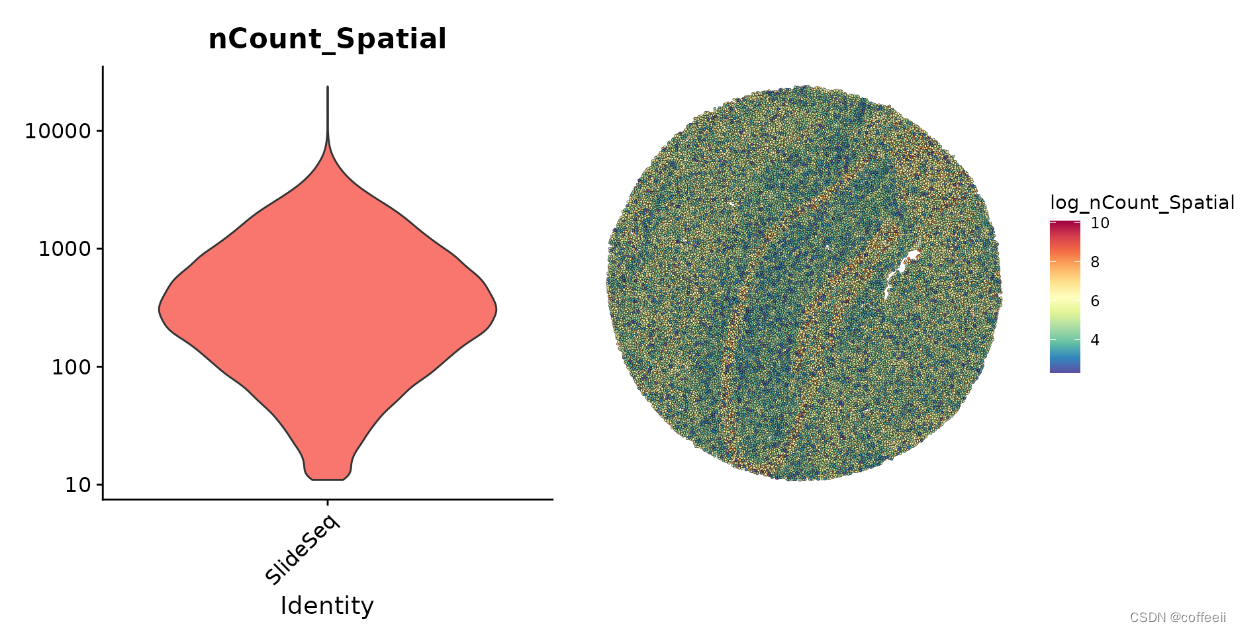
Les étapes de prétraitement initiales des billes pour les données d’expression génique sont similaires à d’autres analyses spatiales de Seurat et à des expériences typiques de scRNA-seq. Ici, nous avons remarqué que de nombreuses billes présentaient un nombre d’UMI particulièrement faible, mais nous avons choisi de conserver toutes les billes détectées pour une analyse en aval.
plot1 <- VlnPlot(slide.seq, features = "nCount_Spatial", pt.size = 0, log = TRUE) + NoLegend()
slide.seq$log_nCount_Spatial <- log(slide.seq$nCount_Spatial)
plot2 <- SpatialFeaturePlot(slide.seq, features = "log_nCount_Spatial") + theme(legend.position = "right")
wrap_plots(plot1, plot2)
Nous avons ensuite utilisé sctransform pour normaliser les données et effectuer des flux de travail standard de réduction de dimensionnalité et de regroupement de scRNA-seq.
slide.seq <- SCTransform(slide.seq, assay = "Spatial", ncells = 3000, verbose = FALSE)
slide.seq <- RunPCA(slide.seq)
slide.seq <- RunUMAP(slide.seq, dims = 1:30)
slide.seq <- FindNeighbors(slide.seq, dims = 1:30)
slide.seq <- FindClusters(slide.seq, resolution = 0.3, verbose = FALSE)
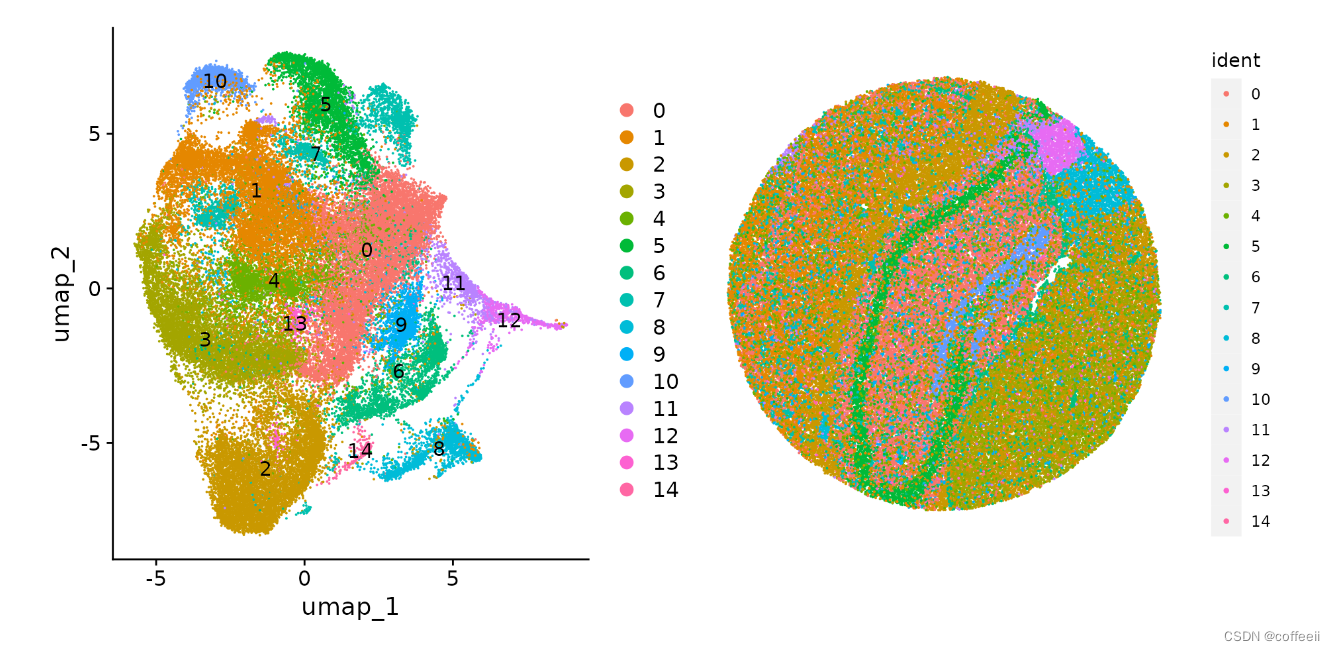
DimPlot()Nous pouvons ensuite visualiser les résultats du clustering dans l'espace UMAP (avec) ou dans l'espace de coordonnées des perles en utilisant SpatialDimPlot().
plot1 <- DimPlot(slide.seq, reduction = "umap", label = TRUE)
plot2 <- SpatialDimPlot(slide.seq, stroke = 0)
plot1 + plot2
SpatialDimPlot(slide.seq, cells.highlight = CellsByIdentities(object = slide.seq, idents = c(1,
6, 13)), facet.highlight = TRUE)

Intégré à la référence scRNA-seq
Pour faciliter l'annotation des types de cellules des ensembles de données Slide-seq, nous exploitons les ensembles de données hippocampiques RNA-seq unicellulaires de souris existants produits par Saunders*, Macosko* et al. 2018. Les données peuvent être téléchargées sous forme d'objets Seurat traités et les matrices de comptage brut sont disponibles sur le site DropViz.
ref <- readRDS("../data/mouse_hippocampus_reference.rds")
ref <- UpdateSeuratObject(ref)
Les annotations originales de l'article sont fournies dans les métadonnées des cellules de l'objet Seurat. Ces annotations sont fournies dans plusieurs « résolutions », allant des grandes catégories ( classe réf.) aux types de cellules (classe réf.) en passant par les types de cellules (réf.c l a ss ) à un sous-cluster au sein d'un type de cellule ( re f sous-cluster). Pour les besoins de cette vignette, nous apporterons une modification à l'annotation du type de cellule ( ref$celltype ), qui, à notre avis, constitue un bon équilibre.
Nous allons d’abord exécuter la méthode de transfert d’étiquettes Seurat pour prédire le type cellulaire prédominant pour chaque bille.
anchors <- FindTransferAnchors(reference = ref, query = slide.seq, normalization.method = "SCT",
npcs = 50)
predictions.assay <- TransferData(anchorset = anchors, refdata = ref$celltype, prediction.assay = TRUE,
weight.reduction = slide.seq[["pca"]], dims = 1:50)
slide.seq[["predictions"]] <- predictions.assay
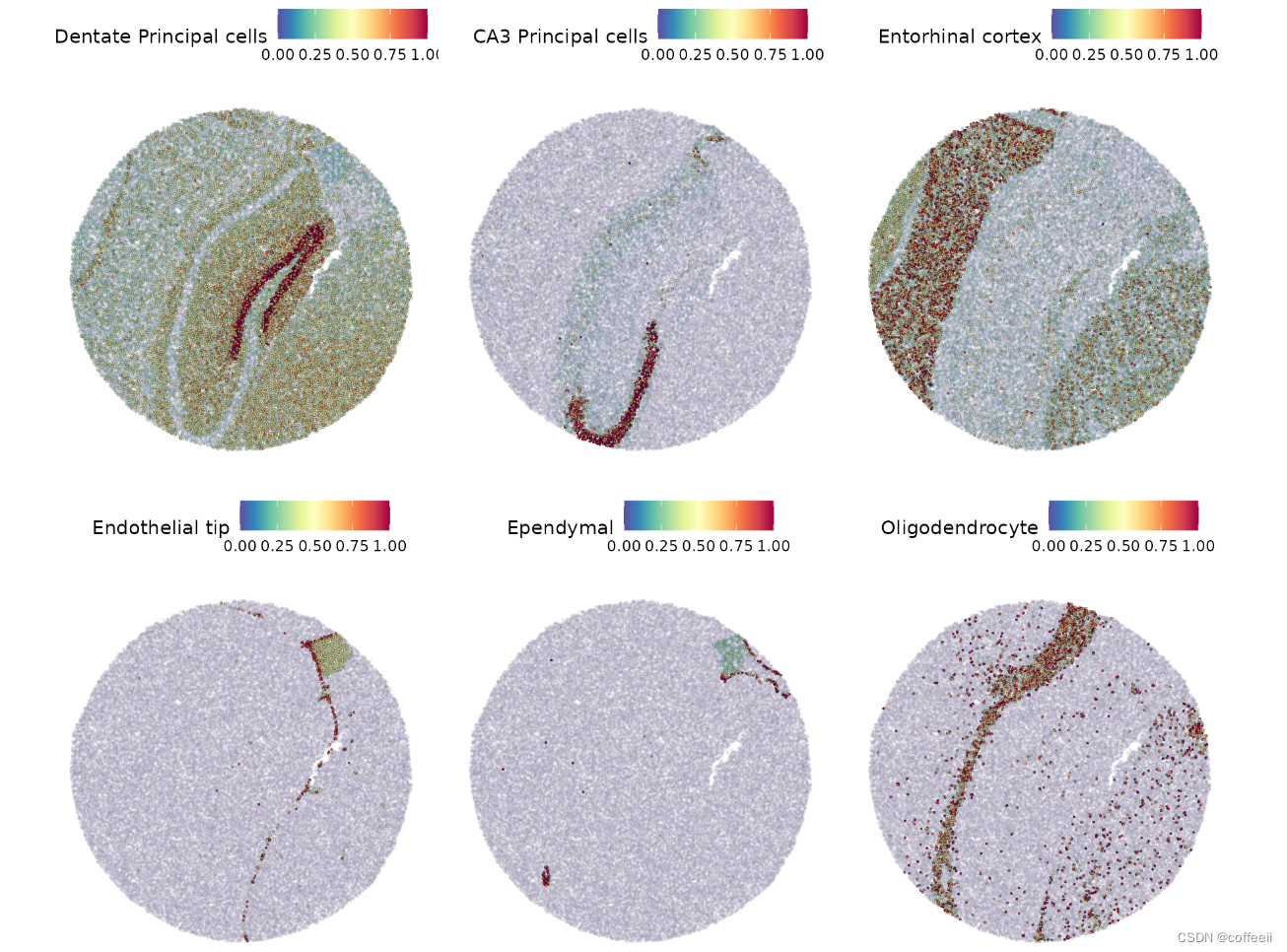
Nous pouvons alors visualiser les scores de prédiction pour certaines des principales catégories attendues.
DefaultAssay(slide.seq) <- "predictions"
SpatialFeaturePlot(slide.seq, features = c("Dentate Principal cells", "CA3 Principal cells", "Entorhinal cortex",
"Endothelial tip", "Ependymal", "Oligodendrocyte"), alpha = c(0.1, 1))
slide.seq$predicted.id <- GetTransferPredictions(slide.seq)
Idents(slide.seq) <- "predicted.id"
SpatialDimPlot(slide.seq, cells.highlight = CellsByIdentities(object = slide.seq, idents = c("CA3 Principal cells",
"Dentate Principal cells", "Endothelial tip")), facet.highlight = TRUE)

Identification d'entités spatialement variables
Comme mentionné dans la vignette Visium, nous pouvons identifier les caractéristiques spatialement variables de deux manières générales : des tests d'expression différentielle entre des régions anatomiques pré-annotées ou des statistiques qui mesurent la dépendance des caractéristiques à l'emplacement spatial.
FindSpatiallyVariableFeatures() Nous démontrons ici ce dernier en définissant une implémentation qui fournit la méthode I de Moran = 'moransi'. Moran's I calcule l'autocorrélation spatiale globale et donne une statistique (similaire au coefficient de corrélation) qui mesure la dépendance de l'entité par rapport à sa position spatiale. Cela nous permet de classer les caractéristiques en fonction de la mesure dans laquelle leur expression varie dans l'espace. Pour faciliter une estimation rapide de cette statistique, nous avons mis en œuvre une stratégie de regroupement de base qui dessine une grille rectangulaire sur un disque Slide-seq et fait la moyenne des caractéristiques et des emplacements dans chaque groupe. Le nombre de compartiments dans les directions x et y est contrôlé respectivement par les paramètres x.cuts et y.cuts. De plus, bien que cela ne soit pas obligatoire, l'installation du package Rfast2 facultatif (install.packages('Rfast2')) réduira considérablement le temps d'exécution grâce à une implémentation plus efficace.
DefaultAssay(slide.seq) <- "SCT"
slide.seq <- FindSpatiallyVariableFeatures(slide.seq, assay = "SCT", slot = "scale.data", features = VariableFeatures(slide.seq)[1:1000],
selection.method = "moransi", x.cuts = 100, y.cuts = 100)
Visualisons maintenant l'expression des 6 premiers traits identifiés par le I de Moran.
SpatialFeaturePlot(slide.seq, features = head(SpatiallyVariableFeatures(slide.seq, selection.method = "moransi"),
6), ncol = 3, alpha = c(0.1, 1), max.cutoff = "q95")
SpatialFeaturePlot(slide.seq, features = head(SpatiallyVariableFeatures(slide.seq, selection.method = "moransi"),
6), ncol = 3, alpha = c(0.1, 1), max.cutoff = "q95")

Déconvolution spatiale à l'aide de RCTD
Alors que FindTransferAnchors peut être utilisé pour intégrer des données au niveau du point à partir d'ensembles de données transcriptomiques spatiales, Seurat v5 inclut également la prise en charge d'une décomposition robuste des types de cellules, une méthode de déconvolution des données au niveau du point à partir d'ensembles de données spatiales lors de la fourniture de références scRNA-seq. Il a été démontré que RCTD annote avec précision les données spatiales provenant de diverses technologies, notamment SLIDE-seq, Visium et la plate-forme spatiale in situ 10x Xenium.
Pour exécuter RCTD, nous installons d'abord spacexr à partir du package GitHub qui implémente RCTD.
devtools::install_github("dmcable/spacexr", build_vignettes = FALSE)
Extrayez les informations de nombre, de cluster et de point des requêtes Seurat et des objets de référence pour construire des objets ReferenceRCT utilisés par DSpatialRNA pour l'annotation.
library(spacexr)
# set up reference
ref <- readRDS("../data/mouse_hippocampus_reference.rds")
ref <- UpdateSeuratObject(ref)
Idents(ref) <- "celltype"
# extract information to pass to the RCTD Reference function
counts <- ref[["RNA"]]$counts
cluster <- as.factor(ref$celltype)
names(cluster) <- colnames(ref)
nUMI <- ref$nCount_RNA
names(nUMI) <- colnames(ref)
reference <- Reference(counts, cluster, nUMI)
# set up query with the RCTD function SpatialRNA
slide.seq <- SeuratData::LoadData("ssHippo")
counts <- slide.seq[["Spatial"]]$counts
coords <- GetTissueCoordinates(slide.seq)
colnames(coords) <- c("x", "y")
coords[is.na(colnames(coords))] <- NULL
query <- SpatialRNA(coords, counts, colSums(counts))
À l'aide de l'objet referenceandquery, nous annotons l'ensemble de données et ajoutons des étiquettes de type de cellule à l'objet de requête Seurat. RCTD se parallélise bien, de sorte que plusieurs cœurs peuvent être spécifiés pour des performances plus rapides.
RCTD <- create.RCTD(query, reference, max_cores = 8)
RCTD <- run.RCTD(RCTD, doublet_mode = "doublet")
slide.seq <- AddMetaData(slide.seq, metadata = RCTD@results$results_df)
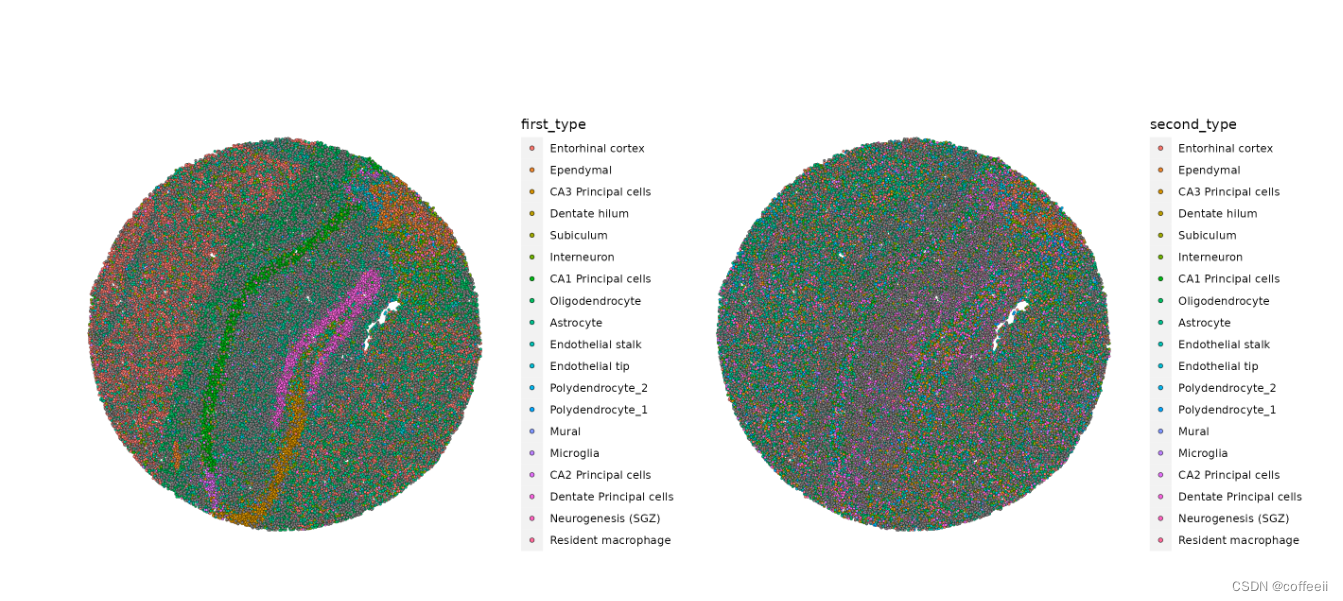
Ensuite, dessinez l'annotation RCTD. Parce que nous exécutons RCTD en mode doublet, l'algorithme attribue une somme first_type à chaque code-barres ou point. second_type
p1 <- SpatialDimPlot(slide.seq, group.by = "first_type")
p2 <- SpatialDimPlot(slide.seq, group.by = "second_type")
p1 | p2
garder
write.csv(x = t(as.data.frame(all_times)), file = "../output/timings/spatial_vignette_times.csv")