CellChat déduit et analyse des informations spatiales pour la communication intercellulaire à partir de données d'imagerie spatiale
Suoqin Jin et Jingren Niu
12 novembre 2022
Chargement des bibliothèques requises
Partie 1 : Saisie, traitement et initialisation des données des objets CellChat
Chargement des données
Création d'un objet CellChat
Configuration de la base de données d'interaction ligand-récepteur
Prétraitement Analyse de la communication cellule à cellule Données d'expression
Partie II : Inférence de réseaux de communication intercellulaires
Calcul des probabilités de communication et déduction des réseaux de communication cellulaires
Déduction de la communication intercellulaire au niveau de la voie de signalisation
Calcul des réseaux de communication intercellulaires agrégés
Partie III : Visualisation des réseaux de communication intercellulaires
Partie V : Enregistrement d'un objet CellChat
Cette vignette décrit les étapes d'utilisation de CellChat pour déduire, analyser et visualiser des réseaux de communication intercellulaires sur un seul ensemble de données d’imagerie spatiale. Nous démontrons que CellChat fonctionne dans les applications dans les données d'imagerie spatiale (1-0-0). Les annotations biologiques (c'est-à-dire les informations sur la population cellulaire) des taches ont été prédites à l'aide de Seurat (//satijalab.org/seurat/articles/spatial_vignette.html).
CellChat nécessite des données d'expression génique et de localisation spatiale des points/cellules comme entrée de l'utilisateur et modélise les cellules en combinant l'expression génique avec une connaissance préalable des distances spatiales et des interactions entre les ligands de signalisation, les récepteurs et la probabilité de communication de leurs cofacteurs.
Après avoir déduit les réseaux de communication intercellulaires, diverses fonctionnalités de CellChat peuvent être utilisées pour une exploration, une analyse et une visualisation plus approfondies des données.
Charger les bibliothèques requises
library(CellChat)
library(patchwork)
options(stringsAsFactors = FALSE)
Partie 1 : Saisie, traitement des données et initialisation de l'objet CellChat
CellChat nécessite quatre entrées utilisateur :
Données d'expression génique pour les spots/cellules : les gènes doivent être disposés en lignes, les noms de lignes et les cellules en colonnes, les noms de colonnes. Des données normalisées (par exemple, taille de bibliothèque normalisée puis transformée en journal à l'aide d'un pseudo-compte de 1) sont requises comme entrée pour l'analyse CellChat. Si l'utilisateur fournit des données de comptage, nous fournissons une fonction normalizeData pour calculer la taille de la bibliothèque, puis effectuons une transformation logarithmique.
Étiquettes de cellules attribuées par l'utilisateur : un bloc de données composé d'informations sur les cellules (les lignes sont des cellules avec des noms de lignes) qui sera utilisé pour définir des groupes de cellules.
Localisation spatiale des spots/cellules : une matrice de données où chaque ligne donne l'emplacement/les coordonnées spatiales de chaque cellule/spot. Pour 10X Visium, ces informations se trouvent dans tissu_positions.csv.
Facteurs d'échelle et diamètres de spot pour les images pleine résolution : contient une liste de facteurs d'échelle et de diamètres de spot pour les images pleine résolution. scale.factors doit contenir un élément nommé spot.diameter, qui est la taille théorique du spot (par exemple, 10x Visium (spot.size = 65 microns)) ; et un autre élément nommé spot, qui est l'image originale en pleine résolution. Le nombre de pixels sur le diamètre théorique du spot. Pour Visium 10X, scale.factors se trouve dans le fichier scalefactors_json.json. le spot est spot.size.fullres.
Télécharger les données
# Here we load a Seurat object of 10X Visium mouse cortex data and its associated cell meta data
load("/Users/jinsuoqin/Mirror/CellChat/tutorial/visium_mouse_cortex_annotated.RData")
library(Seurat)
#> Attaching SeuratObject
visium.brain
#> An object of class Seurat
#> 1298 features across 1073 samples within 3 assays
#> Active assay: SCT (648 features, 260 variable features)
#> 2 other assays present: Spatial, predictions
#> 2 dimensional reductions calculated: pca, umap
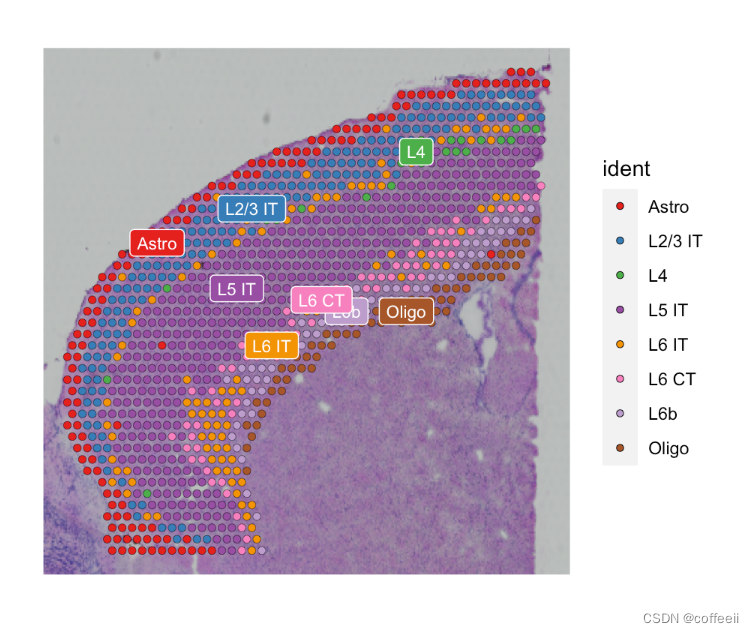
# show the image and annotated spots
SpatialDimPlot(visium.brain, label = T, label.size = 3, cols = scPalette(nlevels(visium.brain)))
#> Scale for 'fill' is already present. Adding another scale for 'fill', which
#> will replace the existing scale.
# Prepare input data for CelChat analysis
data.input = GetAssayData(visium.brain, slot = "data", assay = "SCT") # normalized data matrix
meta = data.frame(labels = Idents(visium.brain), row.names = names(Idents(visium.brain))) # manually create a dataframe consisting of the cell labels
unique(meta$labels) # check the cell labels
#> [1] L2/3 IT Astro L6b L5 IT L6 IT L6 CT L4 Oligo
#> Levels: Astro L2/3 IT L4 L5 IT L6 IT L6 CT L6b Oligo
# load spatial imaging information
# Spatial locations of spots from full (NOT high/low) resolution images are required
spatial.locs = GetTissueCoordinates(visium.brain, scale = NULL, cols = c("imagerow", "imagecol"))
# Scale factors and spot diameters of the full resolution images
scale.factors = jsonlite::fromJSON(txt = file.path("/Users/jinsuoqin/Mirror/CellChat/tutorial/spatial_imaging_data_visium-brain", 'scalefactors_json.json'))
scale.factors = list(spot.diameter = 65, spot = scale.factors$spot_diameter_fullres, # these two information are required
fiducial = scale.factors$fiducial_diameter_fullres, hires = scale.factors$tissue_hires_scalef, lowres = scale.factors$tissue_lowres_scalef # these three information are not required
)
# USER can also extract scale factors from a Seurat object, but the `spot` value here is different from the one in Seurat. Thus, USER still needs to get the `spot` value from the json file.
###### Applying to different types of spatial imaging data ######
# `spot.diameter` is dependent on spatial imaging technologies and `spot` is dependent on specific datasets
Créer un objet CellChat
L'utilisateur peut créer un nouvel objet CellChat à partir d'une matrice de données ou d'un Seurat. Si l'entrée est un objet Seurat, les métadonnées de l'objet seront utilisées par défaut et USER doit fournir group.by pour définir le groupe de cellules. Par exemple, pour l'identité de cellule par défaut dans un objet Seurat, group.by = "ident".
Remarque : Si l'utilisateur charge un objet CellChat précédemment calculé (version < 1.6.0), mettez à jour l'objet via updateCellChat
cellchat <- createCellChat(object = data.input, meta = meta, group.by = "labels",
datatype = "spatial", coordinates = spatial.locs, scale.factors = scale.factors)
#> [1] "Create a CellChat object from a data matrix"
#> Create a CellChat object from spatial imaging data...
#> Set cell identities for the new CellChat object
#> The cell groups used for CellChat analysis are Astro L2/3 IT L4 L5 IT L6 IT L6 CT L6b Oligo
cellchat
#> An object of class CellChat created from a single dataset
#> 648 genes.
#> 1073 cells.
#> CellChat analysis of spatial data! The input spatial locations are
#> imagerow imagecol
#> AAACAGAGCGACTCCT-1 3164 7950
#> AAACCGGGTAGGTACC-1 6517 3407
#> AAACCGTTCGTCCAGG-1 7715 4371
#> AAACTCGTGATATAAG-1 4242 9258
#> AAAGGGATGTAGCAAG-1 4362 5747
#> AAATAACCATACGGGA-1 3164 7537
Mise en place de la base de données d'interaction ligand-récepteur
Notre base de données CellChatDB est une base de données organisée manuellement contenant des interactions ligand-récepteur humain et murin étayées par la littérature. CellChatDB chez la souris contient 2 021 interactions moléculaires validées, dont 60 % d’interactions de signalisation autocrine/paracrine, 21 % d’interactions matrice extracellulaire (ECM)-récepteur et 19 % d’interactions cellule-cellule, interaction de contact. CellChatDB contient 1 939 interactions moléculaires validées chez l'homme, dont 61,8 % d'interactions de signalisation paracrine/autocrine, 21,7 % d'interactions matrice extracellulaire (ECM)-récepteur et 16,5 % d'interactions de contact cellule-cellule.
Les utilisateurs peuvent mettre à jour CellChatDB en ajoutant leurs propres paires ligand-récepteur. Consultez notre tutoriel pour savoir comment procéder.
CellChatDB <- CellChatDB.mouse # use CellChatDB.human if running on human data
# use a subset of CellChatDB for cell-cell communication analysis
CellChatDB.use <- subsetDB(CellChatDB, search = "Secreted Signaling") # use Secreted Signaling
# use all CellChatDB for cell-cell communication analysis
# CellChatDB.use <- CellChatDB # simply use the default CellChatDB
# set the used database in the object
cellchat@DB <- CellChatDB.use
Prétraitement des données d'expression pour l'analyse de la communication intercellulaire
Pour déduire une communication spécifique à l'état cellulaire, nous identifions les ligands ou les récepteurs surexprimés dans une population cellulaire, puis identifions les interactions ligand-récepteur surexprimés lorsque le ligand ou le récepteur est surexprimé.
Nous fournissons également des fonctionnalités permettant de projeter des données d'expression génique sur des réseaux d'interaction protéine-protéine (PPI). Plus précisément, un processus de diffusion est utilisé pour lisser les valeurs d'expression des gènes sur la base des valeurs d'expression des gènes voisins définis dans un réseau protéine-protéine de haute confiance validé expérimentalement. Cette fonctionnalité est utile lors de l’analyse de données unicellulaires avec une faible profondeur de séquençage, car la projection réduit l’effet d’abandon des gènes de signalisation, en particulier pour une éventuelle expression nulle des sous-unités ligand/récepteur. On pourrait s’inquiéter des artefacts que ce processus de diffusion pourrait introduire, mais il n’introduirait que des communications très faibles. Les UTILISATEURS peuvent également ignorer cette étape et définir raw.use = TRUE dans la fonction calculateCommunProb().
# subset the expression data of signaling genes for saving computation cost
cellchat <- subsetData(cellchat) # This step is necessary even if using the whole database
future::plan("multiprocess", workers = 4) # do parallel
#> Warning: Strategy 'multiprocess' is deprecated in future (>= 1.20.0). Instead,
#> explicitly specify either 'multisession' or 'multicore'. In the current R
#> session, 'multiprocess' equals 'multisession'.
#> Warning in supportsMulticoreAndRStudio(...): [ONE-TIME WARNING] Forked
#> processing ('multicore') is not supported when running R from RStudio
#> because it is considered unstable. For more details, how to control forked
#> processing or not, and how to silence this warning in future R sessions, see ?
#> parallelly::supportsMulticore
cellchat <- identifyOverExpressedGenes(cellchat)
cellchat <- identifyOverExpressedInteractions(cellchat)
# project gene expression data onto PPI (Optional: when running it, USER should set `raw.use = FALSE` in the function `computeCommunProb()` in order to use the projected data)
# cellchat <- projectData(cellchat, PPI.mouse)
Partie II : Inférence des réseaux de communication intercellulaires
CellChat déduit une communication de cellule à cellule biologiquement significative en attribuant une valeur de probabilité à chaque interaction et en effectuant des tests de permutation. CellChat modélise les possibilités de communication de cellule à cellule en combinant l'expression des gènes avec la localisation spatiale et les connaissances déjà connues sur les interactions entre les ligands de signalisation, les récepteurs et leurs cofacteurs en utilisant la loi de l'action de masse.
Le nombre de paires ligand-récepteur déduit dépend évidemment de la méthode utilisée pour calculer l’expression génique moyenne pour chaque population cellulaire. En raison de la faible sensibilité des techniques d’imagerie spatiale actuelles, nous recommandons d’utiliser une moyenne tronquée de 10 % pour calculer l’expression moyenne des gènes. L'approche « triméenne » par défaut produit moins d'interactions et peut manquer des signaux de faible expression. Dans ComputeCommunProb, nous offrons la possibilité de calculer l’expression moyenne des gènes à l’aide de différentes méthodes. Il convient de noter que la « trimée » est proche de la moyenne seuil de 25 %, ce qui signifie que si le pourcentage de cellules exprimant dans un groupe est inférieur à 25 %, l'expression moyenne des gènes est nulle. Pour utiliser une moyenne tronquée de 10 %, l'utilisateur peut définir type = "truncatedMean" et trim = 0,1. La fonction calculateAveExpr peut aider à vérifier l'expression moyenne des gènes de signalisation d'intérêt, par exemple, calculateAveExpr(cellchat, Features = c("CXCL12", "CXCR4"), type = "truncatedMean", trim = 0.1).
Calculer les probabilités de communication et déduire les réseaux de communication cellulaire
Pour vérifier rapidement les résultats de l'inférence, les utilisateurs peuvent définir nboot=20 dans calculateCommunProb. Alors « pvalue < 0,05 » signifie qu'aucun des résultats de permutation n'est supérieur à la probabilité de communication observée.
Si une voie de signalisation bien connue dans le processus biologique étudié n'est pas prédite, l'utilisateur peut essayer truncatedMean pour modifier la méthode de calcul de l'expression génique moyenne pour chaque groupe de cellules en utilisant une valeur de trim inférieure.
scale.distance Les utilisateurs peuvent avoir besoin d'ajuster le paramètre lors du traitement des données provenant d'autres techniques d'imagerie spatiale. Veuillez consulter la documentation via ?computeCommunProb pour plus de détails.
cellchat <- computeCommunProb(cellchat, type = "truncatedMean", trim = 0.1,
distance.use = TRUE, interaction.length = 200, scale.distance = 0.01)
#> truncatedMean is used for calculating the average gene expression per cell group.
#> [1] ">>> Run CellChat on spatial imaging data using distances as constraints <<< [2022-11-12 07:49:23]"
#> The suggested minimum value of scaled distances is in [1,2], and the calculated value here is 1.30553
#> [1] ">>> CellChat inference is done. Parameter values are stored in `object@options$parameter` <<< [2022-11-12 08:10:42]"
# Filter out the cell-cell communication if there are only few number of cells in certain cell groups
cellchat <- filterCommunication(cellchat, min.cells = 10)
Déduire une communication de cellule à cellule au niveau de la voie de signalisation
CellChat calcule les probabilités de communication au niveau de la voie de signalisation en additionnant les probabilités de communication pour toutes les interactions ligand-récepteur associées à chaque voie de signalisation.
REMARQUE : Le réseau de communication intercellulaire déduit pour chaque paire ligand-récepteur et chaque voie de signalisation est stocké respectivement dans les emplacements « net » et « netP ».
cellchat <- computeCommunProbPathway(cellchat)
Réseaux de communication de cellule à cellule agrégés informatiquement
Nous pouvons calculer des réseaux de communication intercellulaires agrégés en comptant le nombre de liens ou en résumant les probabilités de communication. sources.useUSER peut également être utilisé pour calculer des cibles de réseau globales.use entre des sous-ensembles de groupes de cellules en définissant la somme de .
cellchat <- aggregateNet(cellchat)
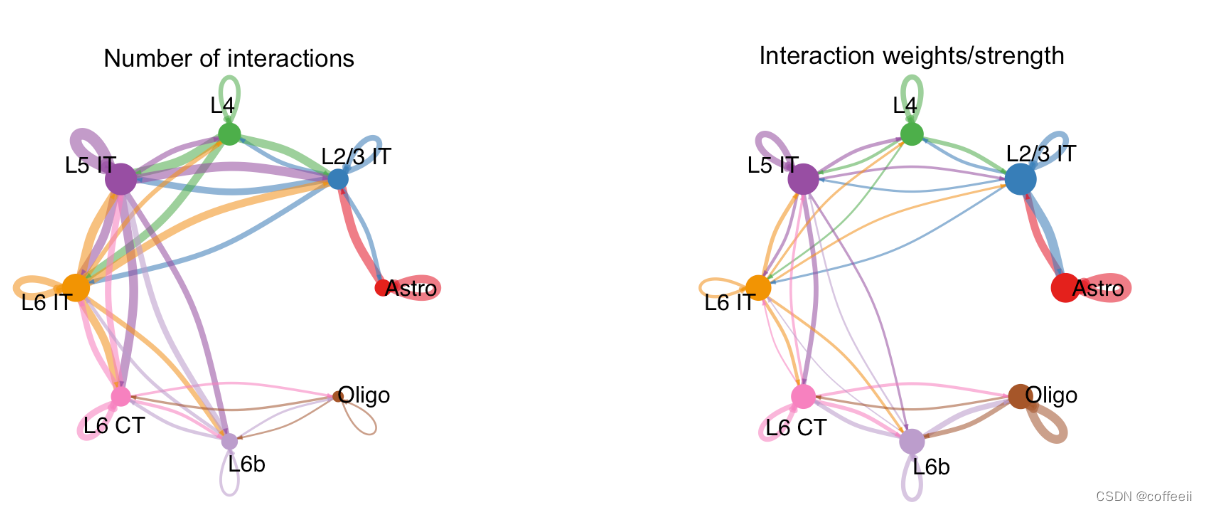
Nous pouvons également visualiser des réseaux de communication intercellulaires agrégés. Par exemple, utilisez un graphique circulaire pour afficher le nombre d’interactions ou la force totale d’interaction (poids) entre deux groupes de cellules.
groupSize <- as.numeric(table(cellchat@idents))
par(mfrow = c(1,2), xpd=TRUE)
netVisual_circle(cellchat@net$count, vertex.weight = rowSums(cellchat@net$count), weight.scale = T, label.edge= F, title.name = "Number of interactions")
netVisual_circle(cellchat@net$weight, vertex.weight = rowSums(cellchat@net$weight), weight.scale = T, label.edge= F, title.name = "Interaction weights/strength")
Partie III : Visualisation des réseaux de communication intercellulaires
Après avoir déduit les réseaux de communication intercellulaires, CellChat offre diverses fonctionnalités pour une exploration, une analyse et une visualisation plus approfondies des données. Ici, nous montrons uniquement le tracé circulaire et le nouveau tracé spatial.
Visualisation de la communication intercellulaire à différents niveaux : vous pouvez utiliser le réseau de communication déduit netVisual_aggregate pour visualiser la voie de signalisation, et utiliser le réseau de communication déduit netVisual_individual pour visualiser les paires LR individuelles liées à la voie de signalisation.
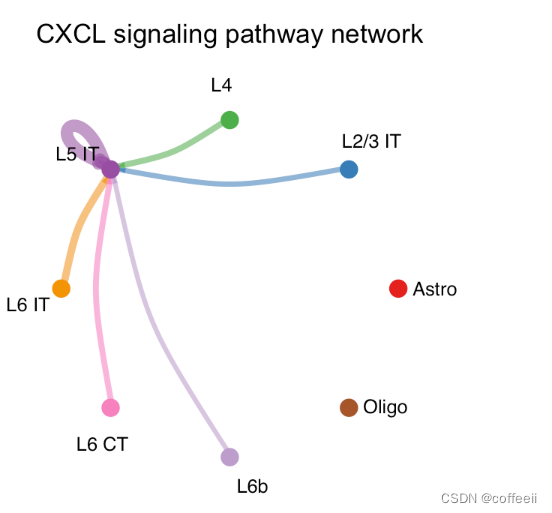
Ici, nous prenons comme exemple l’entrée d’un chemin de signal. Toutes les voies de signalisation montrant les communications importantes sont accessibles à cellchat@netP$pathways.
pathways.show <- c("CXCL")
# Circle plot
par(mfrow=c(1,1))
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "circle")
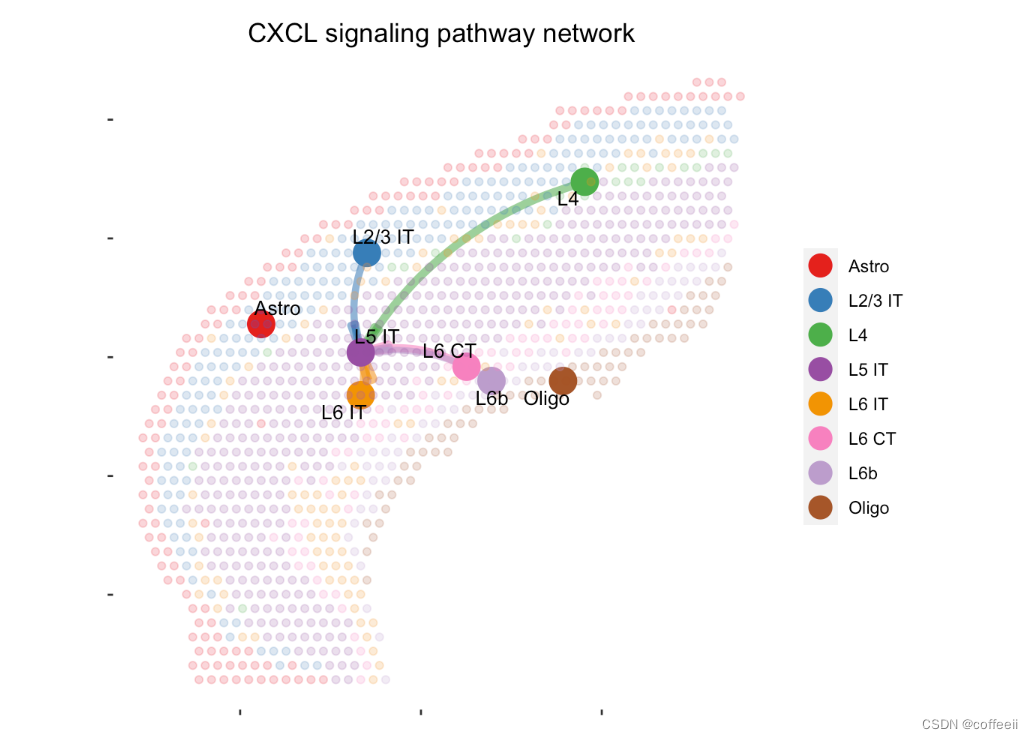
# Spatial plot
par(mfrow=c(1,1))
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "spatial", edge.width.max = 2, vertex.size.max = 1, alpha.image = 0.2, vertex.label.cex = 3.5)
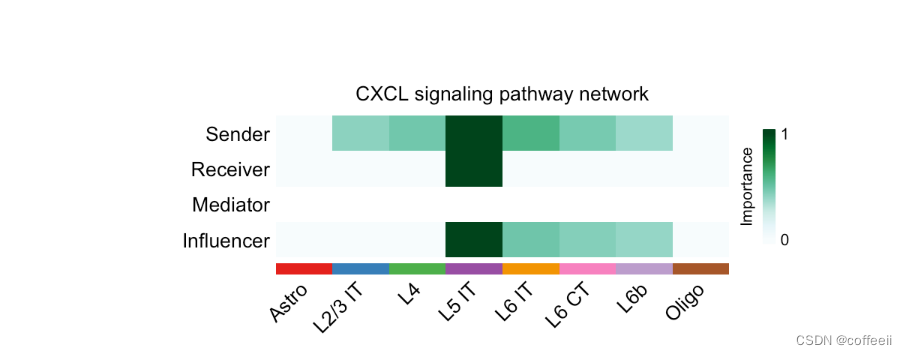
Calculez et visualisez les scores de centralité du réseau :
# Compute the network centrality scores
cellchat <- netAnalysis_computeCentrality(cellchat, slot.name = "netP") # the slot 'netP' means the inferred intercellular communication network of signaling pathways
# Visualize the computed centrality scores using heatmap, allowing ready identification of major signaling roles of cell groups
par(mfrow=c(1,1))
netAnalysis_signalingRole_network(cellchat, signaling = pathways.show, width = 8, height = 2.5, font.size = 10)
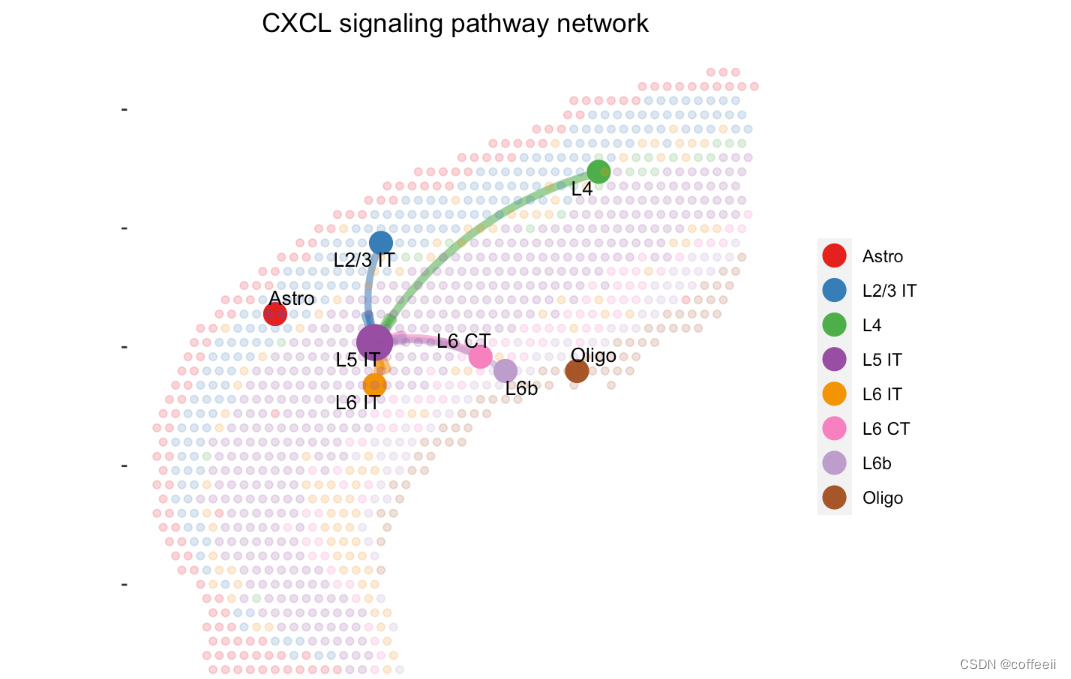
# USER can visualize this information on the spatial imaging, e.g., bigger circle indicates larger incoming signaling
par(mfrow=c(1,1))
netVisual_aggregate(cellchat, signaling = pathways.show, layout = "spatial", edge.width.max = 2, alpha.image = 0.2, vertex.weight = "incoming", vertex.size.max = 3, vertex.label.cex = 3.5)
REMARQUE : Les réseaux de communication de cellule à cellule sont déduits des données d’imagerie spatiale, et diverses fonctions de CellChat sont disponibles pour une exploration, une analyse et une visualisation plus approfondies des données. Veuillez vérifier les autres fonctionnalités dans le didacticiel de base appelé CellChat-vignette.html
Partie 5 : Enregistrer l'objet CellChat
saveRDS(cellchat, file = "cellchat_visium_mouse_cortex.rds")
sessionInfo()
#> R version 4.1.2 (2021-11-01)
#> Platform: x86_64-apple-darwin17.0 (64-bit)
#> Running under: macOS Big Sur 10.16
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRlapack.dylib
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] SeuratObject_4.0.4 Seurat_4.0.6 patchwork_1.1.1
#> [4] CellChat_1.6.0 Biobase_2.54.0 BiocGenerics_0.40.0
#> [7] ggplot2_3.3.5 igraph_1.3.4 dplyr_1.0.7
#>
#> loaded via a namespace (and not attached):
#> [1] backports_1.4.1 circlize_0.4.13 systemfonts_1.0.2
#> [4] NMF_0.23.0 plyr_1.8.6 lazyeval_0.2.2
#> [7] splines_4.1.2 BiocParallel_1.28.3 listenv_0.8.0
#> [10] scattermore_0.7 ggnetwork_0.5.10 gridBase_0.4-7
#> [13] digest_0.6.29 foreach_1.5.1 htmltools_0.5.2
#> [16] magick_2.7.3 ggalluvial_0.12.3 fansi_0.5.0
#> [19] magrittr_2.0.1 tensor_1.5 cluster_2.1.2
#> [22] doParallel_1.0.16 ROCR_1.0-11 sna_2.6
#> [25] ComplexHeatmap_2.10.0 globals_0.14.0 matrixStats_0.61.0
#> [28] svglite_2.0.0 spatstat.sparse_2.1-0 colorspace_2.0-2
#> [31] ggrepel_0.9.1 xfun_0.33 crayon_1.4.2
#> [34] jsonlite_1.7.2 spatstat.data_2.1-2 survival_3.2-13
#> [37] zoo_1.8-9 iterators_1.0.13 glue_1.6.0
#> [40] polyclip_1.10-0 registry_0.5-1 gtable_0.3.0
#> [43] leiden_0.3.9 GetoptLong_1.0.5 car_3.0-12
#> [46] future.apply_1.8.1 shape_1.4.6 abind_1.4-5
#> [49] scales_1.1.1 DBI_1.1.2 rngtools_1.5.2
#> [52] rstatix_0.7.0 miniUI_0.1.1.1 Rcpp_1.0.7
#> [55] viridisLite_0.4.0 xtable_1.8-4 clue_0.3-60
#> [58] spatstat.core_2.3-2 reticulate_1.22 stats4_4.1.2
#> [61] htmlwidgets_1.5.4 httr_1.4.2 FNN_1.1.3
#> [64] RColorBrewer_1.1-2 ellipsis_0.3.2 ica_1.0-2
#> [67] farver_2.1.0 pkgconfig_2.0.3 uwot_0.1.11
#> [70] deldir_1.0-6 sass_0.4.0 utf8_1.2.2
#> [73] labeling_0.4.2 later_1.3.0 tidyselect_1.1.1
#> [76] rlang_0.4.12 reshape2_1.4.4 munsell_0.5.0
#> [79] tools_4.1.2 generics_0.1.1 statnet.common_4.5.0
#> [82] broom_0.7.10 ggridges_0.5.3 evaluate_0.17
#> [85] stringr_1.4.0 fastmap_1.1.0 goftest_1.2-3
#> [88] yaml_2.2.1 knitr_1.40 fitdistrplus_1.1-6
#> [91] purrr_0.3.4 RANN_2.6.1 nlme_3.1-153
#> [94] pbapply_1.5-0 future_1.23.0 mime_0.12
#> [97] compiler_4.1.2 plotly_4.10.0 png_0.1-7
#> [100] ggsignif_0.6.3 spatstat.utils_2.3-0 tibble_3.1.6
#> [103] bslib_0.3.1 stringi_1.7.6 highr_0.9
#> [106] RSpectra_0.16-0 lattice_0.20-45 Matrix_1.3-4
#> [109] vctrs_0.3.8 pillar_1.6.4 lifecycle_1.0.1
#> [112] spatstat.geom_2.3-1 lmtest_0.9-39 jquerylib_0.1.4
#> [115] GlobalOptions_0.1.2 RcppAnnoy_0.0.19 BiocNeighbors_1.12.0
#> [118] data.table_1.14.2 cowplot_1.1.1 irlba_2.3.5
#> [121] httpuv_1.6.4 R6_2.5.1 promises_1.2.0.1
#> [124] network_1.17.1 gridExtra_2.3 KernSmooth_2.23-20
#> [127] IRanges_2.28.0 parallelly_1.30.0 codetools_0.2-18
#> [130] MASS_7.3-54 assertthat_0.2.1 pkgmaker_0.32.2
#> [133] rjson_0.2.20 withr_2.4.3 sctransform_0.3.2
#> [136] S4Vectors_0.32.3 mgcv_1.8-38 parallel_4.1.2
#> [139] rpart_4.1-15 grid_4.1.2 tidyr_1.1.4
#> [142] coda_0.19-4 rmarkdown_2.17 carData_3.0-4
#> [145] Rtsne_0.15 ggpubr_0.4.0 shiny_1.7.1