Anmerkung des Herausgebers: Die Forschung und Entwicklung von KI-Arzneimitteln ist eine der wichtigen Richtungen für die zukünftige Anwendung künstlicher Intelligenz. Seit dem ersten Ausbruch des neuen Coronavirus (SARS-CoV-2) hat die Forschung und Entwicklung niedermolekularer Medikamente gegen das neue Coronavirus große Aufmerksamkeit erregt, und der erste kürzlich abgehaltene Wettbewerb für KI-Arzneimittelentwicklungsalgorithmen konzentrierte sich darauf. Im Wettbewerb erzielte das Team vom Scientific Intelligence Center von Microsoft Research hervorragende Ergebnisse und gewann mit seinen innovativen KI-Modellsystemen AI2BMD und ViSNet die Meisterschaft.
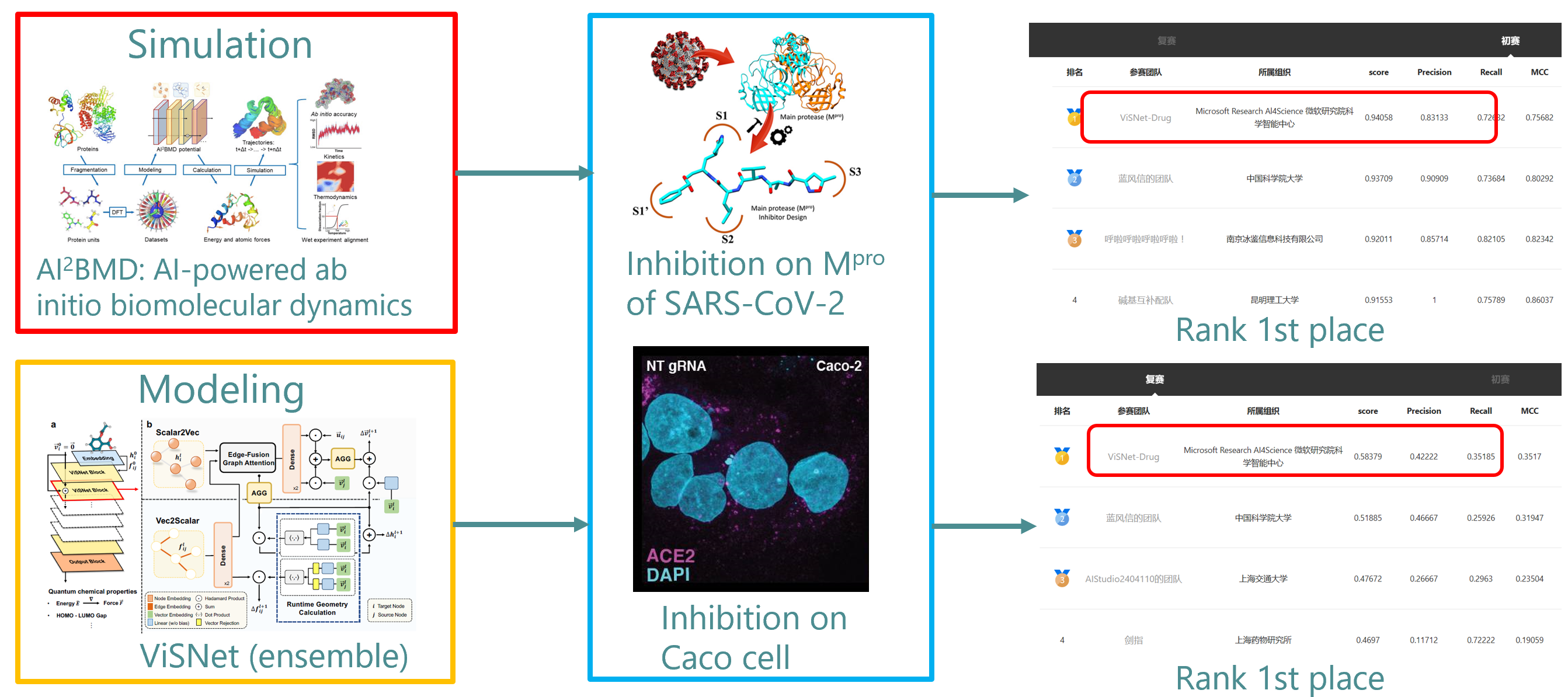
Kürzlich wurden die Ergebnisse des ersten AI Drug Research and Development Algorithm Competition bekannt gegeben, der gemeinsam von der Tsinghua University School of Pharmacy, Baidu Paddle, Baidu Smart Cloud und Lingang Laboratory organisiert wurde. Ein Team des Scientific Intelligence Center von Microsoft Research nutzte den Quantenpräzisionsleistung, entwickelt von dem wissenschaftlichen Simulationssystem AI2BMD und dem universellen molekularen dreidimensionalen Strukturnetzwerk ViSNet, belegte in der Vorrunde, im Halbfinale und im Finale den ersten Platz und gewann die Gesamtmeisterschaft des Wettbewerbs, was das Anwendungspotenzial von KI in demonstrierte Förderung der Arzneimittelforschung und -entwicklung.

Das Team des Microsoft Research Scientific Intelligence Center gewann den ersten Wettbewerb für KI-Arzneimittelentwicklungsalgorithmen
Dieser Wettbewerb wird von branchenführenden Organisationen wie der Chinese Pharmaceutical Association vollständig unterstützt. Insgesamt nehmen 878 Teams aus der ganzen Welt teil. Als globale technologische Innovationsveranstaltung konzentriert sich dieser Wettbewerb auf die Forschung und Entwicklung niedermolekularer Medikamente gegen das neue Coronavirus (SARS-CoV-2). Tatsächlich hat die Entwicklung niedermolekularer Medikamente gegen das neue Coronavirus seit dem ersten Ausbruch des neuen Coronavirus große Aufmerksamkeit erregt. Um die Ausbreitung von COVID-19 zu bekämpfen, ist ein tiefes Verständnis der Replikations- und Infektionsmechanismen des Virus von entscheidender Bedeutung. Unter ihnen ist die neue Coronavirus-Hauptprotease (Mpro) ein Schlüsselenzym, das dafür verantwortlich ist, die vom Virus während des Infektionsprozesses produzierten Proteinvorläufer zu schneiden und die Virusreplikation zu fördern. Daher ist die Hauptprotease ein potenzielles therapeutisches Ziel. Die Hemmung ihrer Aktivität kann wirksam in den Virusreplikationsprozess eingreifen und damit einen Durchbruch für Behandlungsmethoden bedeuten. Daher müssen die Teilnehmer in der Vorphase dieses Wettbewerbs mithilfe von Deep Learning, molekularem Docking und anderen Methoden eine Modellierung durchführen, um die Wahrscheinlichkeit vorherzusagen, dass kleine Moleküle die Aktivität der Hauptprotease hemmen. Im Halbfinale wird es um die Wahrscheinlichkeit gehen kleine Moleküle, die die Replikation des neuen Coronavirus auf Caco-Zellen hemmen. .
In der Vorrunde der Arzneimittelvorhersage für die Hauptprotease des neuen Coronavirus sah sich das Team des Microsoft Research Scientific Intelligence Center mit dem Problem konfrontiert, dass häufig verwendete molekulare Docking-Software nicht effektiv zwischen positiven und negativen Proben und der freien Bindungsenergie von Zielproteinen unterscheiden kann verwendeten das neu entwickelte AI2BMD-Simulationssystem [1] und verbesserten so die Genauigkeit der Arzneimittelvorhersage erheblich. Das AI2BMD-Simulationssystem ermöglicht genaue Berechnungen verschiedener Proteinenergien und -kräfte über 10.000 Atome und ist breit anwendbar. Im Vergleich zur Dichtefunktionaltheorie (DFT) verkürzt sich die Berechnungszeit des AI2BMD-Simulationssystems um mehrere Größenordnungen. Mit Dynamiksimulationen von Hunderten von Nanosekunden demonstriert AI2BMD seine herausragenden Fähigkeiten bei der Erforschung des Proteinkonformationsraums, der Vorhersage experimenteller NMR-Daten und der Simulation von Proteinfaltungsprozessen. Im Vergleich zu herkömmlichen molekularen Docking- und klassischen Dynamiksimulationsmethoden bietet das AI2BMD-System auch offensichtliche Vorteile bei der Berechnung der freien Bindungsenergie.
Link zum Papier zum AI2BMD-Simulationssystem: https://www.biorxiv.org/content/10.1101/2023.07.12.548519v1
Im Halbfinale nutzte das Team das selbst entwickelte geometrische Deep-Learning-Modell ViSNet für die molekulare Modellierung [2], um Repräsentationslernen an zusammengesetzten Molekülen durchzuführen. ViSNet ist eine potenzielle Funktion für maschinelles Lernen im AI2BMD-Simulationssystem. Als äquivariantes geometrisch verbessertes graphisches neuronales Netzwerk kann ViSNet geometrische Merkmale (Abstände, Winkel, Diederwinkel usw.) mit der Komplexität linearer Berechnungen extrahieren. ViSNet übertrifft andere hochmoderne Methoden bei mehreren Molekulardynamik-Benchmarks, darunter MD17, rMD17 und MD22, und erzielt gleichzeitig hervorragende Vorhersagen quantenchemischer Eigenschaften in QM9- und Molecule3D-Datensätzen.
Im Halbfinale nutzte das Team außerdem den unabhängig entwickelten ersten selbst entwickelten vollständigen Konformationsraumdatensatz für Proteinmakromoleküle AIMD-Chig [3] und den öffentlichen Datensatz für kleine Moleküle OGB, um die dreidimensionale Strukturdarstellung von Proteinen vorab zu trainieren bzw. kleine Moleküle, und dann wird das Modell durch Multitasking-Lernen verfeinert. Diese Methode erzielte nicht nur die beste Vorhersagegenauigkeit, sondern führte auch das zweitplatzierte Team im Wettbewerb mit großem Vorsprung an. In der abschließenden Verteidigung erreichte die Algorithmuslösung zur Vorhersage von COVID-19-Medikamenten des Microsoft Research Scientific Intelligence Center-Teams eine hervorragende Gesamtpunktzahl von 99,60 Punkten, die deutlich über den Endergebnissen von 90,76 Punkten für den Zweitplatzierten und 85,31 Punkten lag für den dritten zweiten Platz im Wettbewerb.
Der neue Algorithmus zur Vorhersage von Coronavirus-Medikamenten, vorgeschlagen vom Team des Microsoft Research Scientific Intelligence Center
Durch diesen Arzneimittelforschungs- und -entwicklungswettbewerb zeigte das vom Scientific Intelligence Center von Microsoft Research entwickelte Quantenpräzisionsdynamik-Simulationssystem AI2BMD ein hervorragendes praktisches Anwendungspotenzial. Es wird erwartet, dass AI2BMD in Zukunft umfassendere Untersuchungen zur Erklärung molekularer Mechanismen von Lebensaktivitäten, Arzneimitteldesign, Enzymkatalyse usw. durchführen und dazu beitragen wird, die Entwicklung der KI-Arzneimittelforschung und -entwicklung zu beschleunigen.
[1] Wang T, He X, Li M, et al. AI2BMD: Effiziente Charakterisierung der Proteindynamik mit Ab-initio-Genauigkeit. bioRxiv, 2023: 2023.07. 12.548519.
[2] Wang Y, Li S, Wang T, et al. ViSNet: ein skalierbares und genaues geometrisches Deep-Learning-Potenzial für die Simulation der Molekulardynamik. arXiv-Vorabdruck arXiv:2210.16518, 2022.
[3] Wang T, He X, Li M, et al. AIMD-Chig: Erforschung des Konformationsraums eines 166 Atome umfassenden Proteins Chignolin mit Ab-initio-Molekulardynamik. Sci Data 10, 549 (2023).