En los últimos años, con el desarrollo de la tecnología de secuenciación de alto rendimiento, la secuenciación del transcriptoma se ha convertido en el principal medio para estudiar la regulación de la expresión génica. Sabemos que las transcripciones de muchas especies son muy diversas y complejas, y la mayoría de los genes eucariotas no se ajustan al modelo de "un gen, una transcripción", y estos genes a menudo tienen múltiples formas empalmadas. A través de la secuenciación de próxima generación, la expresión génica y la investigación cuantitativa se pueden llevar a cabo con mucha precisión, pero debido a la limitación de la longitud de lectura, no se puede obtener la información de la transcripción completa. Por lo tanto, el transcriptoma de longitud completa basado en la plataforma de secuenciación de tercera generación se ha convertido en un nuevo auge de la investigación.
El transcriptoma de longitud completa (Full-length transcriptoma) se basa en la plataforma de secuenciación de tres generaciones PacBio y Nanopore, sin interrumpir el empalme, obtiene directamente la secuencia de ARNm de longitud completa y la información de estructura completa, incluidos 5'UTR, 3'UTR y poliA cola, para analizar con precisión Hay información estructural como genes de fusión y empalme alternativos de especies con genomas de referencia, lo que supera los problemas de empalme de transcripción corta e información incompleta de especies sin genomas de referencia. Al mismo tiempo, con la ayuda de los datos de secuenciación de próxima generación, se puede realizar un análisis de expresión específico de la transcripción para obtener información de anotación más completa.
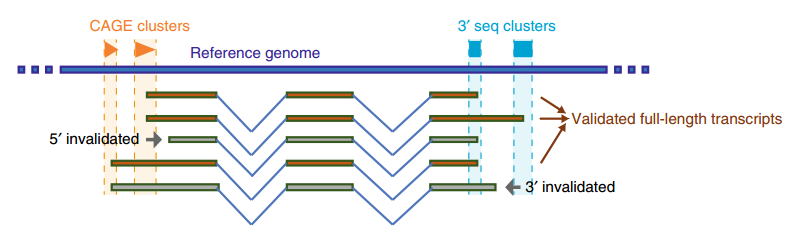
Figura 1 Transcripciones completas confirmadas en ratas
Principios de la secuenciación del transcriptoma de longitud completa
proceso de experimento
Shi YanLiuCh

Figura 2 Proceso de experimento de transcriptoma de longitud completa
Secuenciación CCS (secuenciación de círculo rodante)
PistolaHuanCeXu
El transcriptoma de longitud completa utiliza tecnología de secuenciación en tiempo real de una sola molécula para construir una biblioteca en forma de mancuerna y secuenciarla en ciclo de manera circular. Es más probable que las bibliotecas de fragmentos cortos caigan en guías de ondas de modo cero (ZMW) durante la secuenciación.
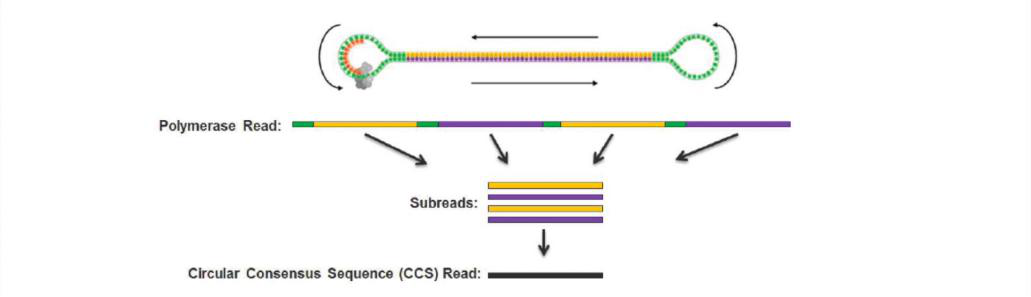
Figura 3 Secuenciación CCS del transcriptoma de longitud completa
-
Lectura de polimerasa: la secuencia leída directamente por la secuenciación de tercera generación, incluidos los adaptadores y las inserciones;
-
Sublecturas: retire el adaptador de lectura de polimerasa y genere una sublectura cada vez que se pruebe el fragmento de inserción;
-
Lectura CCS: la secuencia de consenso generada por sublecturas (al menos dos) derivadas de la misma lectura de polimerasa.
Diseño de proyecto de transcripción completa
Para las especies con genomas de referencia , la información completa del transcriptoma puede corregir el ensamblaje incorrecto del genoma, descubrir con mayor precisión nuevos transcritos y genes, y analizar los eventos de fusión de genes.
Para las especies con genomas de referencia incompletos , el transcriptoma de longitud completa puede optimizar la estructura del gen, asistir en el ensamblaje y la anotación del genoma, y mejorar la precisión de la expresión génica y la utilización de datos.
Para las especies sin un genoma de referencia , la biblioteca de especies Unigene se construye mediante la secuenciación del transcriptoma de longitud completa de tres generaciones, y la secuencia de referencia en el nivel del transcriptoma de la especie (genoma de referencia en el nivel del transcriptoma) se puede obtener sin ensamblar la secuencia, siempre que una gran cantidad de apoyo para la investigación posterior Buena base de información genética.
El transcriptoma tiene especificidad espacio-temporal, y el diseño experimental se realiza de acuerdo al propósito de la investigación, así:
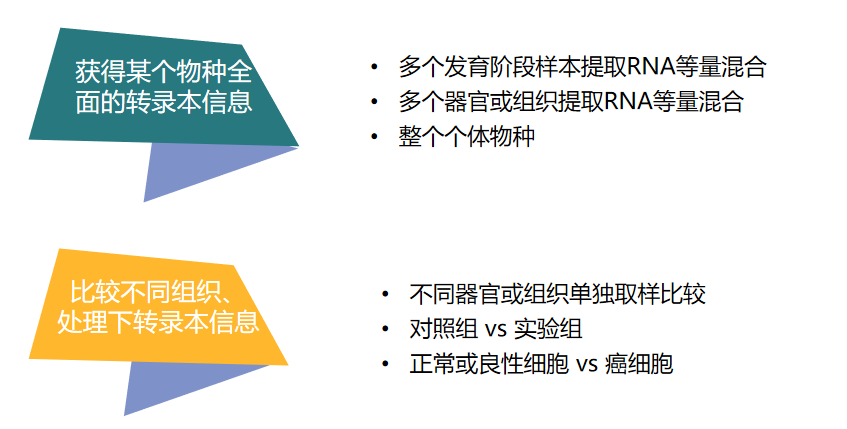
Figura 4 Diseño experimental de transcriptoma completo

Se recomienda utilizar 2+3 generaciones de tecnología de secuenciación de transcriptomas al mismo tiempo para garantizar la precisión estructural, la integridad de la secuencia y la precisión de la expresión de la secuencia, para lograr el uso óptimo de los datos y el rendimiento de costo más alto. De acuerdo con los diferentes requisitos y propósitos, para la mayoría de las especies, la cantidad recomendada de datos de secuenciación es de 8 a 12 G. Para estudiar genes de baja expresión, eventos de corte y empalme variables, poliploides o especies con una gran cantidad de genes, es necesario aumentar la cantidad de datos de secuenciación. cantidad de datos adecuadamente.
Flujo de trabajo de análisis de transcriptoma completo
Luego de obtener la secuencia de secuenciación original (Sequenced Reads), el análisis bioinformático se realiza mediante el siguiente proceso:
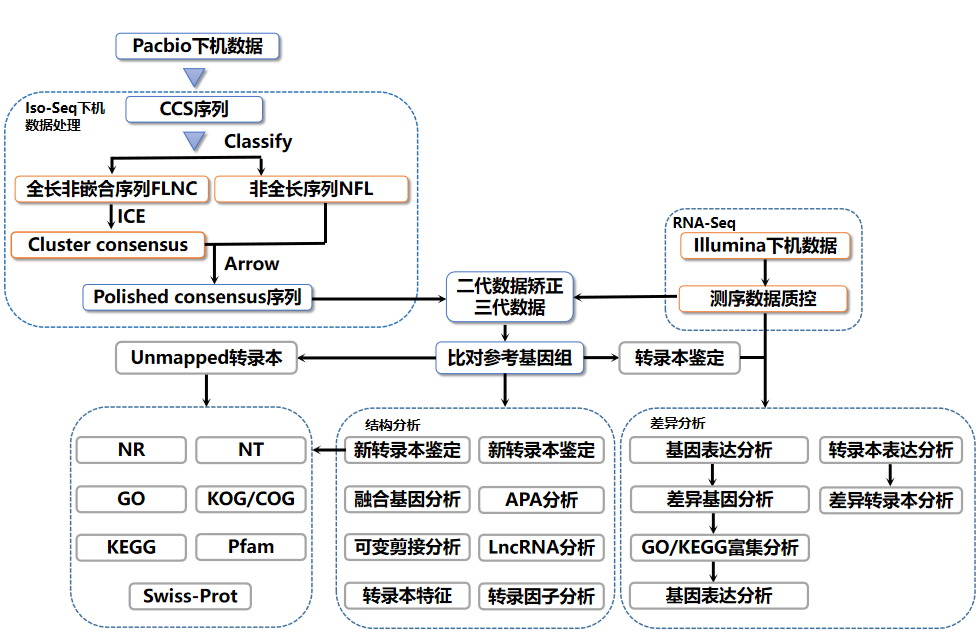
Figura 5 Proceso de análisis bioinformático del transcriptoma completo de Lingen Biotech
Ventajas de la secuenciación del transcriptoma completo de Lingen Bio
-
Sin necesidad de corte y empalme de transcritos, la secuencia de transcritos de longitud completa se puede obtener directamente, lo que refleja de manera más fiel la información del transcriptoma de las especies secuenciadas;
-
Capaz de detectar múltiples formas de empalme alternativas, descubrir más sitios de empalme y eventos de empalme alternativos;
-
Capacidad para descubrir nuevos genes funcionales y complementar las anotaciones del genoma;
-
Contribuye al análisis preciso de genes de fusión, genes homólogos, genes de superfamilia o alelos;
-
Equipo profesional de análisis de bioinformática con amplia experiencia en proyectos.
Solicitud de muestra
-
Requisitos de muestra de ARN total
Generalmente, ARN total > 5 ug/biblioteca, cantidad total > 15 ug (3 bibliotecas), concentración ≧ 250 ng/ul.
-
Requisitos de calidad de la muestra de ARN
OD260/280≧1,8, OD260/230≧1,0, pico normal a 260 nm, valor RIN de ARN total≧8,0, 28S/18S≧1,3.
-
evitar daños en el ADNc
Se utilizan tintes seguros para evitar daños en el ADNc causados por EB y la radiación ultravioleta.
En resumen , el transcriptoma de longitud completa de tres generaciones es aplicable tanto a las especies con ginseng como a las especies sin ginseng, y se pueden obtener más eventos reguladores de la transcripción, lo que refleja más fielmente la información del transcriptoma de las especies secuenciadas.
En la actualidad , la secuenciación completa del transcriptoma para estudiar la estructura del gen se ha convertido en una tendencia de publicación de artículos. Las especies de investigación incluyen sorgo, maíz, Arabidopsis, pollo, humano y ratón, bambú, algodón, etc. ¡Lingen Biotech proporciona servicios profesionales de análisis y secuenciación de transcriptomas completos para ayudarlo en su investigación!

Haga un buen trabajo de secuenciación con corazón y confíe cada resultado