前列腺癌是威胁全球男性的主要癌症之一,存在显著的肿瘤异质性,产生耐药也不尽相同。近年来的研究揭示了其SPOP突变亚型的作用机制,并由此给出一些建设性的治疗建议。
前列腺癌是威胁全球男性的主要癌症之一,且影响其发生发展的因素复杂多样,存在显著的肿瘤异质性,产生耐药也不尽相同。在前列腺癌基因突变样本中,SPOP基因点突变存在于在大约10%。然而BET蛋白是近年来抗肿瘤药物设计的热门靶点,和SPOP有什么关系?SPOP突变是如何导致耐药的?针对这一类型的前列腺癌如何进行分子靶向治疗?本篇研究将会给出一些建设性的答案。
研究动态
该研究首次发现BRD2、BRD3和BRD4(BET蛋白,一类可以和乙酰化组蛋白结合的表观遗传学调控蛋白)是SPOP的作用底物。正常细胞中SPOP通过蛋白酶体途径促进BET蛋白的泛素化降解,将BET蛋白维持在较低水平。
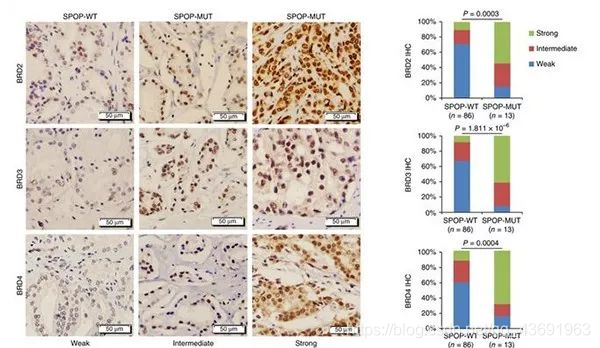
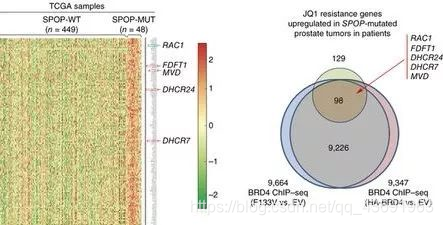
SPOP突变导致其与BET蛋白的相互作用及其促进BET蛋白泛素化降解的能力大为降低,BET蛋白在肿瘤组织中大量积累。BET蛋白如果积累则促进胆固醇合成相关代谢酶类(如FDFT1, DHCR24,DHCR7等)和小GTP酶 RAC1的转录,进而激活AKT/mTORC1信号通路,促进耐药肿瘤细胞的恶性增殖。
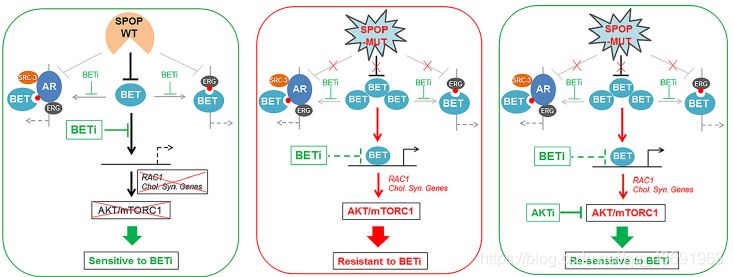
BET小分子抑制剂(如JQ1、I-BET 762见图3)具有广谱、高效的抗肿瘤效果,但单独在突变型SPOP肿瘤细胞中,由于BET蛋白大量积累,所以其抑瘤效果大大降低。然而,和AKT抑制剂联用,则又可以恢复前列腺癌细胞对BET抑制剂的敏感性。
该研究不仅阐明了SPOP突变促进肿瘤恶性增殖的分子机制(F133残疾突变),同时揭示了BET蛋白是SPOP的真正降解底物,SPOP突变亚型前列腺癌对BET抑制剂存在天然耐药现象,并找到了SPOP突变肿瘤恶性增值的相关通路:AKT/mTORC1信号通路。提出了治疗SPOP突变前列腺癌的治疗方法:采用BET抑制剂和AKT抑制剂联合使用能有效逆转BET抑制剂耐药。
M君有话说:
本文的核心意义不仅在于提出BET蛋白可以作为前列腺癌有希望的生物标志物,并且为该亚型前列腺癌的精准治疗提供了理论指导。
参考文献:
[1] P Zhang, et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT–mTORC1 activation. Nature Medicine, 2017, 23, 1055-1062.
>>>>
相关产品
I-BET 762是一种 BET bromodomain 抑制剂,IC50 为 32.5-42.5 nM。