animalcules是一个R软件包,用于利用最新的数据分析、可视化方法和机器学习模型,为用户提供一个易于使用的互动式微生物组分析框架。它可以作为一个独立的软件包使用,或者用户可以用附带的交互式R Shiny应用程序探索他们的数据。
传统的微生物组分析,如α/β多样性和差异丰度分析得到加强,而新的方法,如生物标志物识别,则由动物分子引入。animalcules生成的强大的互动和动态数字使用户能够更好地理解他们的数据并发现新的见解。

介绍
animalcules 包专注于微生物组数据的统一框架,然后包装目前比较流行的分析方法和手段,作者说传统的alpha和beta多样性已经太泛滥了,于是将生物标记物分析的方法引入*animalcules *包中,然后使用交互式的图表来分析和了解数据,更为强大。大家一定要注意,这类框架都使用的S4类对象,不然不会如此方便。
以下教程主要参考软件帮助手册,原文详见:https://bioconductor.org/packages/release/bioc/vignettes/animalcules/inst/doc/animalcules.html
R包安装
软件依赖关系众多,如有些自动安装失败的包,还需要在RStudio中Packages管理中心手动安装。
#--安装BiocManager用于安装Bioconductor的R包
if (!requireNamespace("BiocManager", quietly=TRUE))
install.packages("BiocManager")
#--BiocManager安装Bioconductor中的animalcules包
if (!requireNamespace("animalcules", quietly=TRUE))
BiocManager::install("compbiomed/animalcules")
## 安装github版本
if (!requireNamespace("devtools", quietly=TRUE))
install.packages("devtools")
if (!requireNamespace("animalcules", quietly=TRUE))
devtools::install_github("compbiomed/animalcules")
#下面演示构造MultiAssayExperiment对象时,用到了里面内置的phyloseq对象数据集
if (!requireNamespace("ggClusterNet", quietly=TRUE))
devtools::install_github("taowenmicro/ggClusterNet")
#--载入R包
library(animalcules)
library(SummarizedExperiment)
library(MultiAssayExperiment)
data_dir = system.file("extdata/MAE.rds", package = "animalcules")
MAE = readRDS(data_dir)关于数据格式的必要了解
总之一句话,想要更方便,就要多封装,一致的格式,统一的格式。
作者默认数据解析MAE:其实核心就是SummarizedExperiment数据,多个SummarizedExperiment就是MultiAssayExperiment。这可是多组学数据整合的一个比较好的框架呀。下面我们简单介绍一下
多组学数据处理思路
组学实验越来越普遍、为实验设计,数据整合和分析增加了复杂性。R和Bioconductor为统计分析和可视化提供了一个通用框架,为各种高通量数据类型提供了专门的数据类,但缺乏对多组学实验进行整合分析的方法。
MultiAssayExperiment
多组学实验的整合R包,MultiAssayExperiment,为多种多样的基因组数据提供一致的表示,存储和操作。
MultiAssayExperiment(https://bioconductor.org/packages/MultiAssayExperiment)引入了一个面向对象的S4类对象,定义了用于表示多组学实验的通用数据结构。它有三个关键组成部分:
(i)colData,一个包含患者或细胞系水平的特征(如病理学和组织学)的“主要”数据集;
(ii)ExperimentList,主要数据存储对象,可以包括多组的数据。
(iii)sampleMap,用于全部数据之间联系起来的map文件。
-
(1)构造函数和相关的有效性检查,简化创建MultiAssayExperiment对象,同时允许灵活地表示复杂的实验。
-
(2)允许进行数据选择的子集操作。
MultiAssayExperiment核心数据基于SummarizedExperiment,同时支持异质性的多组学数据。MultiAssayExperiment类和方法提供了一个灵活的框架,用于整合和分析重叠样本的互补分析。它集成了任何支持基本子集和维度名称的数据类,因此默认情况下支持许多数据类,而不需要额外的调整。
#-关于A MultiAssayExperiment object对象的一些基础操作
#assays部分
# assays(MAE)# 提取数据矩阵部分文件,这是一个list,所以提取每个矩阵需要继续
#--下面提取第一个矩阵,这有什么用呢,类似多组学数据,用于操作更加简便。
# assays(MAE)[[1]]# 第二个对象类似
# assays(MAE)[[2]]
#--colData,也就是对数据矩阵列名的注释信息,类似于phyloseq对象中的map文件
colData(MAE)
#--查看子对象数量,都是s4类对象,可以单独提取
#--就单个的S4类对象进行各部分数据的提取
microbe <- MAE[["MicrobeGenetics"]]
otu_table <- as.data.frame(SummarizedExperiment::assays(microbe))
tax_table <- as.data.frame(SummarizedExperiment::rowData(microbe))
map <- as.data.frame(SummarizedExperiment::colData(microbe))构造MultiAssayExperiment对象
使用phyloseq对象构建MultiAssayExperiment对象
至于如何构造phyloseq文件,可以点击查看。想要具体学习什么是phyloseq对象,可以点击
# 如何构建这个MAE对象呢?
#--得到phyloserq对象并提取必要数据信息
library(ggClusterNet)
library(phyloseq)
ps
otu = as.data.frame(t(vegan_otu(ps)))
head(otu)
tax = as.data.frame((vegan_tax(ps)))
head(tax)
map = sample_data(ps)
head(map)
#--首先构造SummarizedExperiment对象,比较简单,类似phyloseq对象
micro <- SummarizedExperiment(assays=list(counts=as.matrix(otu)),
colData=map,
rowData=tax)
# 将SummarizedExperiment对象封装成为ExperimentList
mlist <- ExperimentList()
mlist[[1]] = micro
names(mlist) = "MicrobeGenetics"# 注意必须命名,否则无法区分每个部分数据组
# 构造不同数据组之间的记录文件
gistmap <- data.frame(
primary = row.names(map),
colname = row.names(map),
stringsAsFactors = FALSE)
maplistowe <- list(MicrobeGenetics = gistmap)
sampMapowe <- listToMap(maplistowe)
# colData文件为分组文件,数据框即可,本案例只有一个微生物组数据,所以直接用map文件就可以了。
#-下面就直接构建了MultiAssayExperiment文件
mae <- MultiAssayExperiment(experiments = mlist, colData = map,
sampleMap = sampMapowe)运行Shiny app
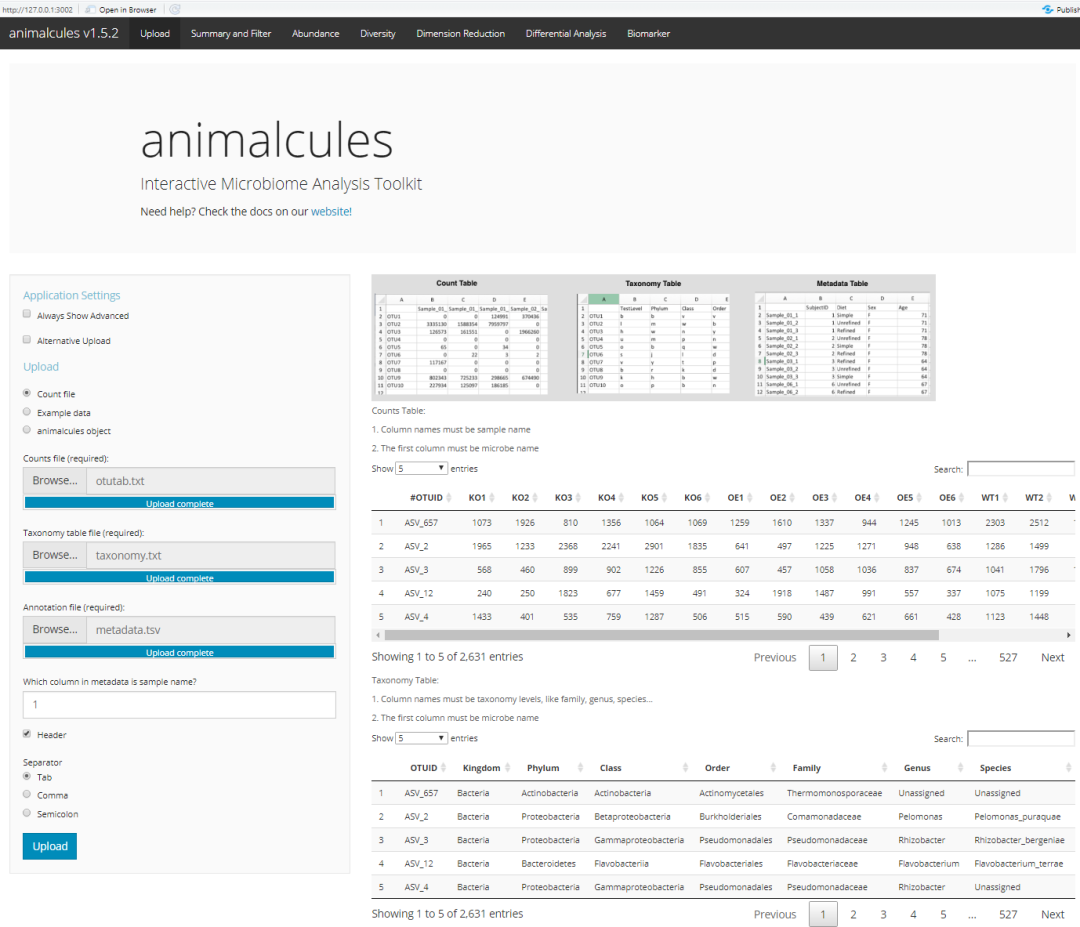
这里我们准备了自己的示例文件,也是为保证尽可能的将这个工具变成一种普通工具,供大家使用。
run_animalcules()### 基础统计
往下的部分,我会同时运行Shiny和官方代码教程,并作比对,但是不会深入挖掘,仅仅会帮助大家完成官方教程和提出一些建议。其次虽然我们也构建MAE文件,但是由于作者代码有一些小瑕疵,所以使用官方数据做演示。
这部分用于统计每个样本中OTU的数量,并做两种方式可视化:频率曲线,箱线+散点图;如果使用shiny程序的话,直接可以展示表格。

此外,可以按照微生物分类水平合并OTU数据:

- samples_discard : 需要去除样本的id
```{R}
# ?filter_summary_pie_box
p <- filter_summary_pie_box(MAE,
samples_discard = c("subject_2", "subject_4"),
filter_type = "By Metadata",
sample_condition = "AGE")
p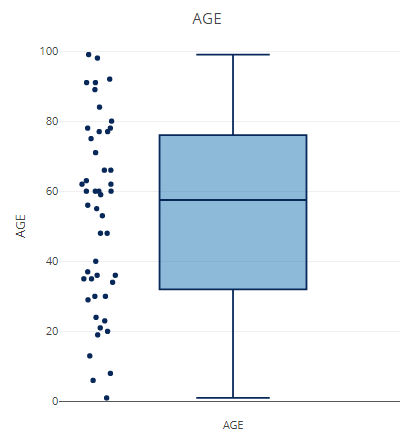
更换分组,重新统计。
p <- filter_summary_bar_density(MAE,
samples_discard = c("subject_2", "subject_4"),
filter_type = "By Metadata",
sample_condition = "SEX")
p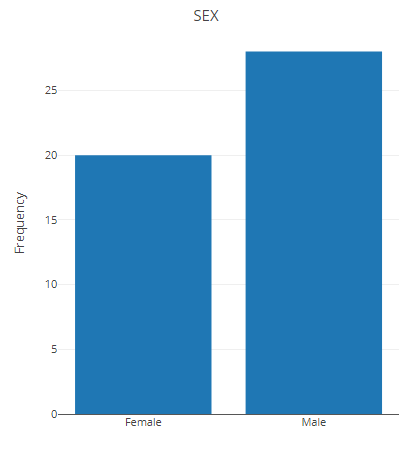
#--提取子集,并且提取map文件
microbe <- MAE[['MicrobeGenetics']]
samples <- as.data.frame(colData(microbe))
result <- filter_categorize(samples,
sample_condition="AGE",
new_label="AGE_GROUP",
bin_breaks=c(0,55,75,100),
bin_labels=c('Young','Adult',"Elderly"))
head(result$sam_table)result$plot.binned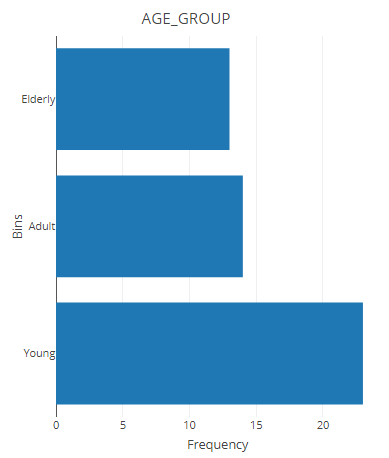
丰度展示-堆叠柱状图
通过tax_level选择某个分类等级,通过 sample_conditions 选择需要添加的分组标签。值得注意的是这里可以对堆叠柱状图排序的,通过order_organisms来指定,默认丰度从高到低。这里从源代码来看就是通过改变factor来实现的,所以图例第一个也是排序的这个微生物。
p <- relabu_barplot(MAE,
tax_level="family",
order_organisms=c('Retroviridae'),
sort_by="organisms",
sample_conditions=c('SEX', 'AGE'),
show_legend=TRUE)
p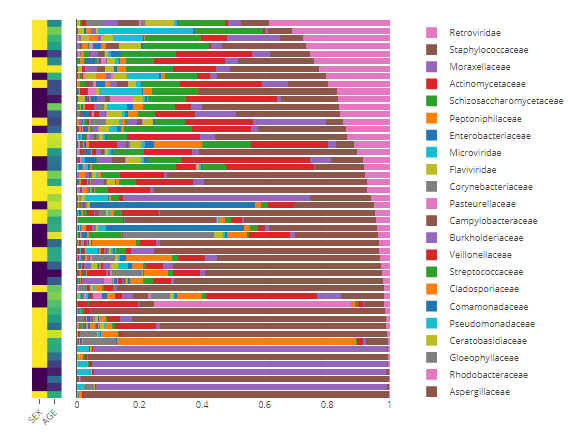
shiny版本:
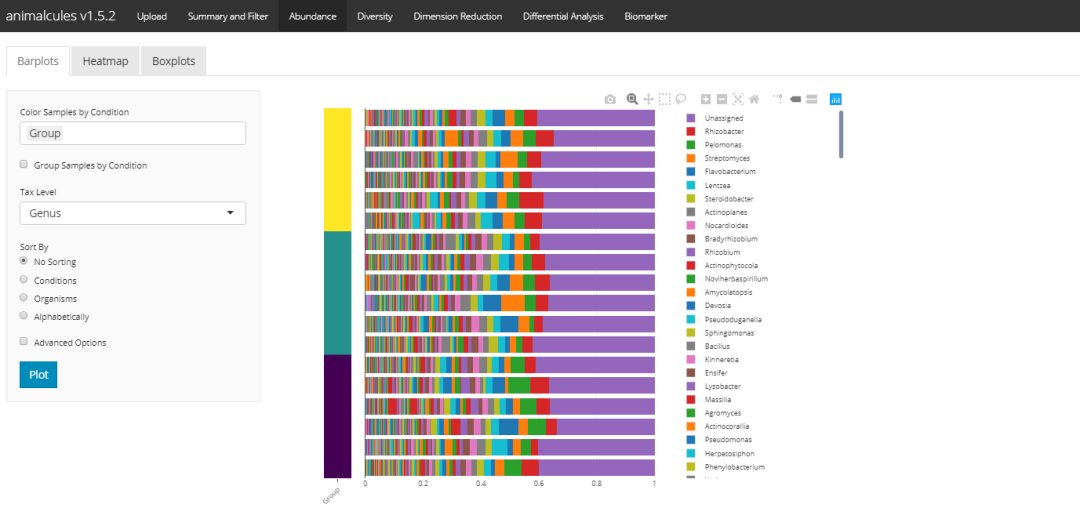
丰度-热图
p <- relabu_heatmap(MAE,
tax_level="genus",
sort_by="conditions",
sample_conditions=c("SEX", "AGE"))
p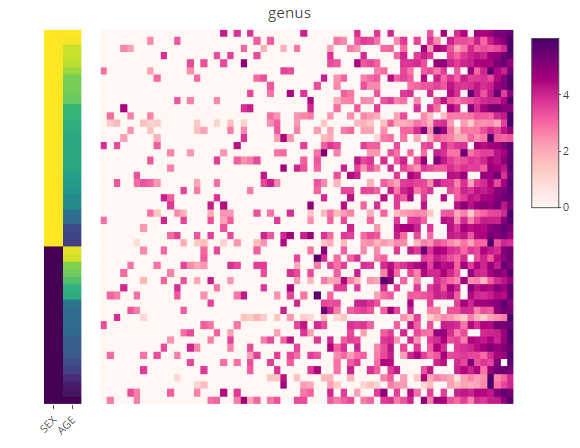
shiny版本:
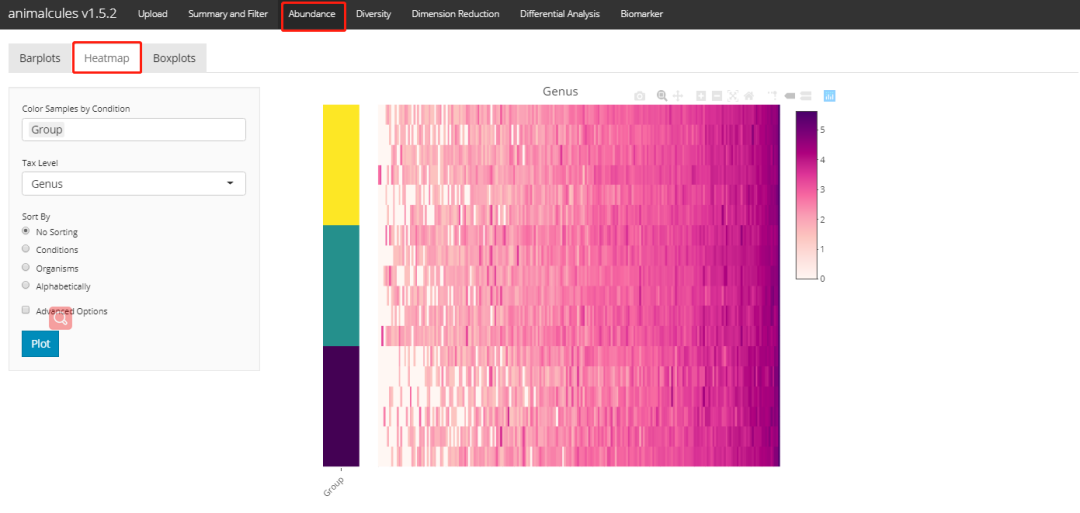
丰度-箱线图
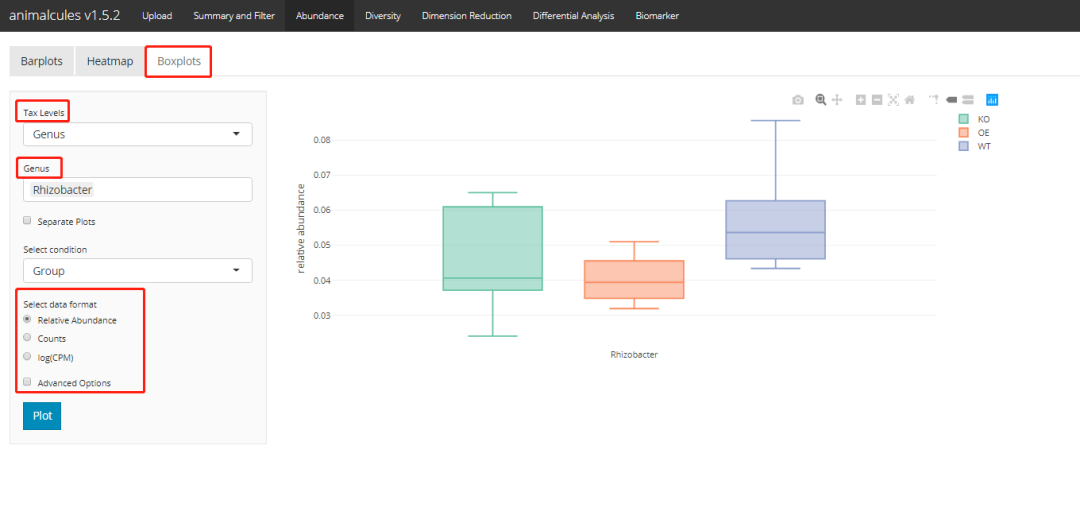
p <- relabu_boxplot(MAE,
tax_level="genus",
organisms=c("Escherichia", "Actinomyces"),
condition="SEX",
datatype="logcpm")
p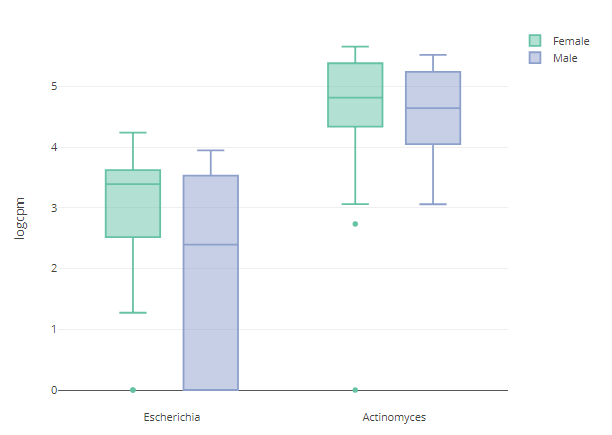
shiny版本:
Alpha多样性
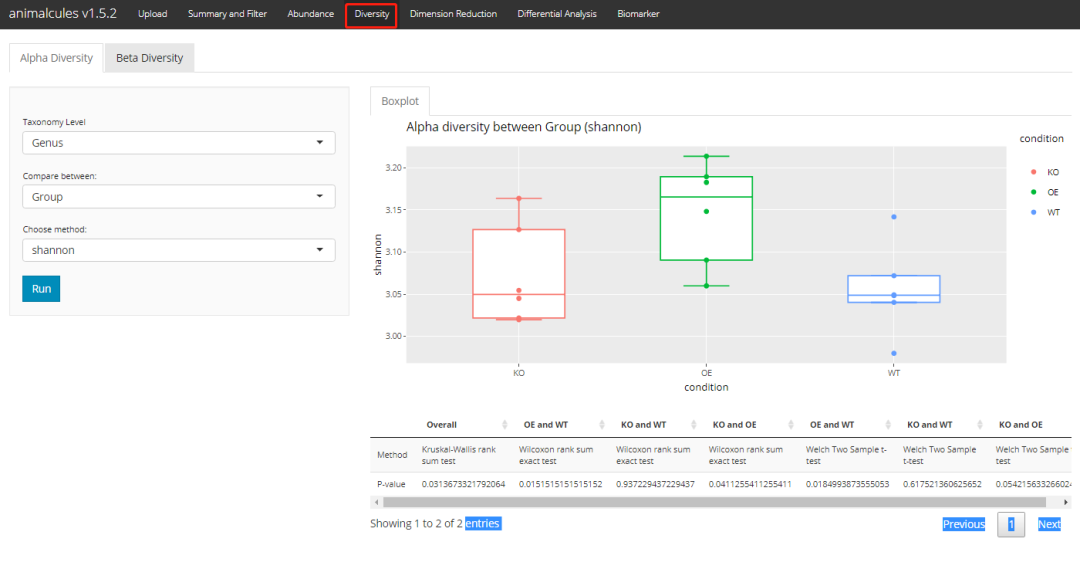
这里只有四个多样性指标。然后通过箱线+散点图展示。
alpha_div_boxplot(MAE = MAE,
tax_level = "genus",
condition = "DISEASE",
alpha_metric = "shannon")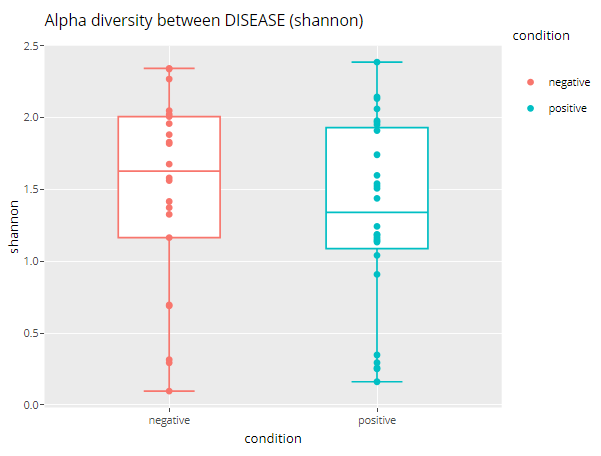
对多样性进行统计检验。这里可选的是”Wilcoxon rank sum test”, “T-test”, “Kruskal-Wallis”这三种方法。
# ?do_alpha_div_test
do_alpha_div_test(MAE = MAE,
tax_level = "genus",
condition = "DISEASE",
alpha_metric = "shannon",
alpha_stat = "T-test")shiny版本:
Beta多样性-聚类距离热图
diversity_beta_heatmap(MAE = MAE,
tax_level = 'genus',
input_beta_method = "bray",
input_bdhm_select_conditions = 'DISEASE',
input_bdhm_sort_by = 'condition')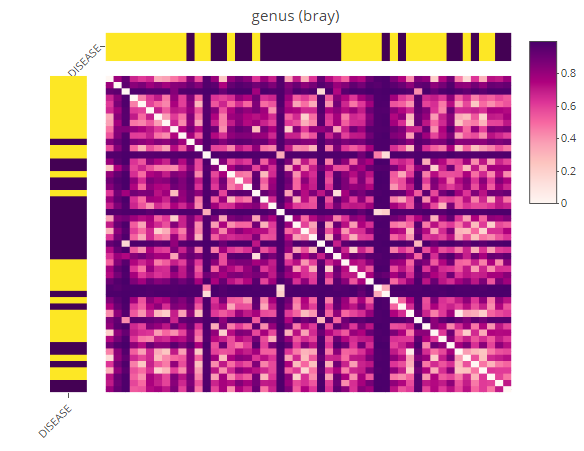
Beta多样性-组间距离箱线图
其次通过组内距离和组间距离的箱线图展示
diversity_beta_boxplot(MAE = MAE,
tax_level = 'genus',
input_beta_method = "bray",
input_select_beta_condition = 'DISEASE')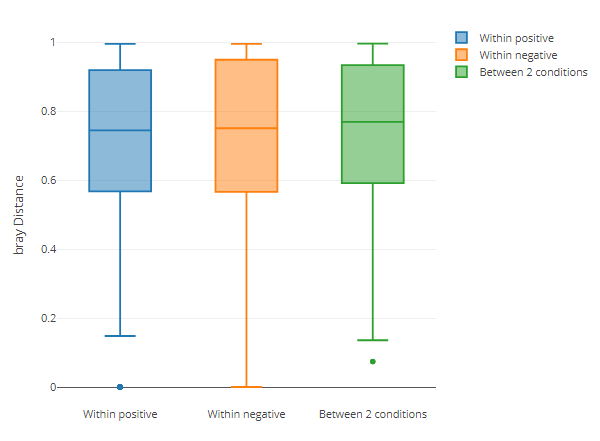
再有就是统计检验,共有三种方法可以选择:PERMANOVA,Kruskal-Wallis,Wilcoxon test。
但是只有两种距离可供选择,其次就是两两比较不能实现。
# ?diversity_beta_test
diversity_beta_test(MAE = MAE,
tax_level = 'genus',
input_beta_method = "bray",
input_select_beta_condition = 'DISEASE',
input_select_beta_stat_method = 'PERMANOVA',
input_num_permutation_permanova = 999)shiny版本:
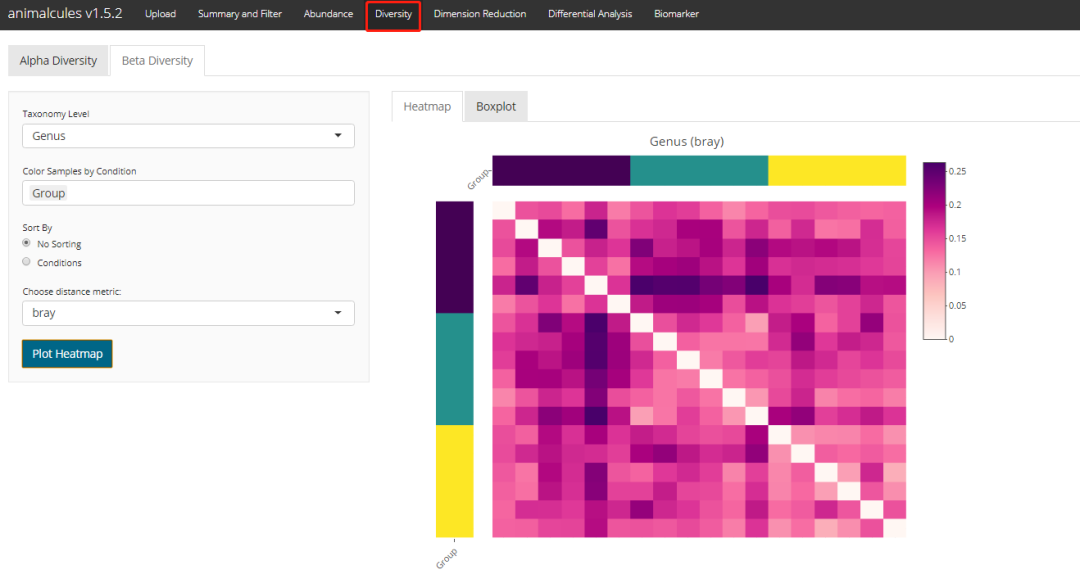
shiny版本(error):
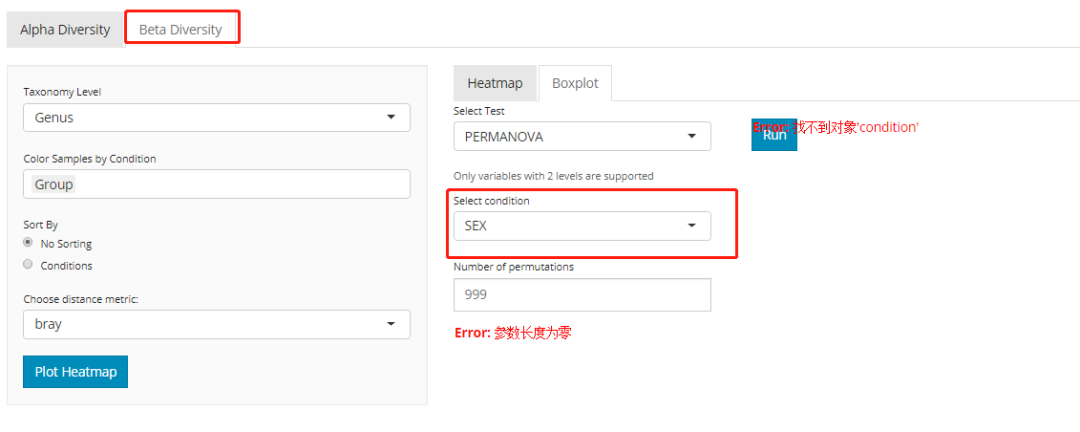
排序
PCA
result <- dimred_pca(MAE,
tax_level="genus",
color="AGE",
shape="DISEASE",
pcx=1,
pcy=2,
datatype="logcpm")
result$plot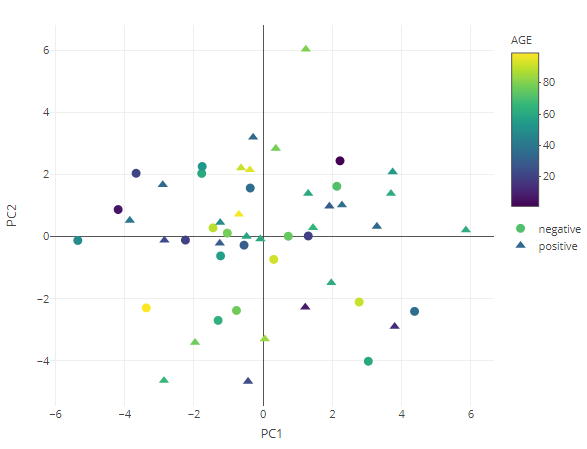
PCoA
result <- dimred_pcoa(MAE,
tax_level="genus",
color="AGE",
shape="DISEASE",
axx=1,
axy=2,
method="bray")
result$plotresult$plot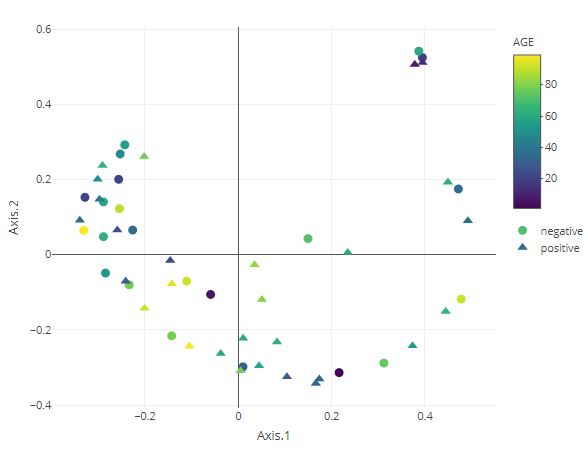
UMAP
result <- dimred_umap(MAE,
tax_level="genus",
color="AGE",
shape="DISEASE",
cx=1,
cy=2,
n_neighbors=15,
metric="euclidean",
datatype="logcpm")
result$plot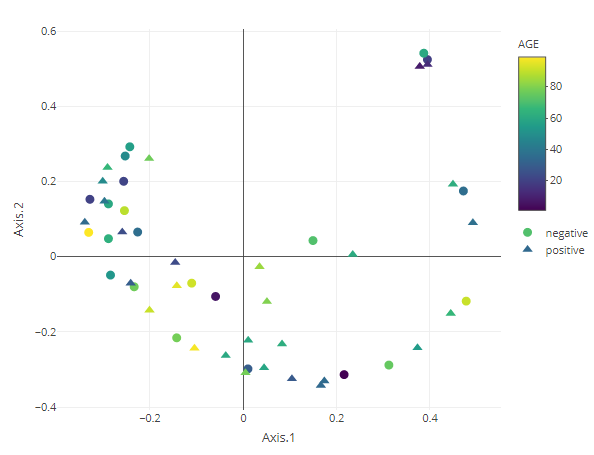
t-SNE
除了二维图形展示还可以进行三维图形的展示。
result <- dimred_tsne(MAE,
tax_level="phylum",
color="AGE",
shape="GROUP",
k="3D",
initial_dims=30,
perplexity=10,
datatype="logcpm")
result$plot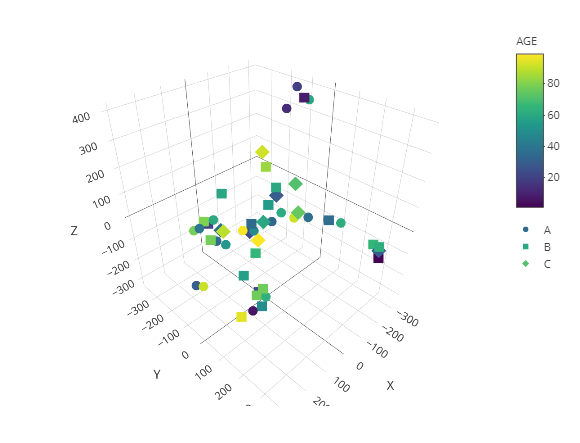
shiny版本:
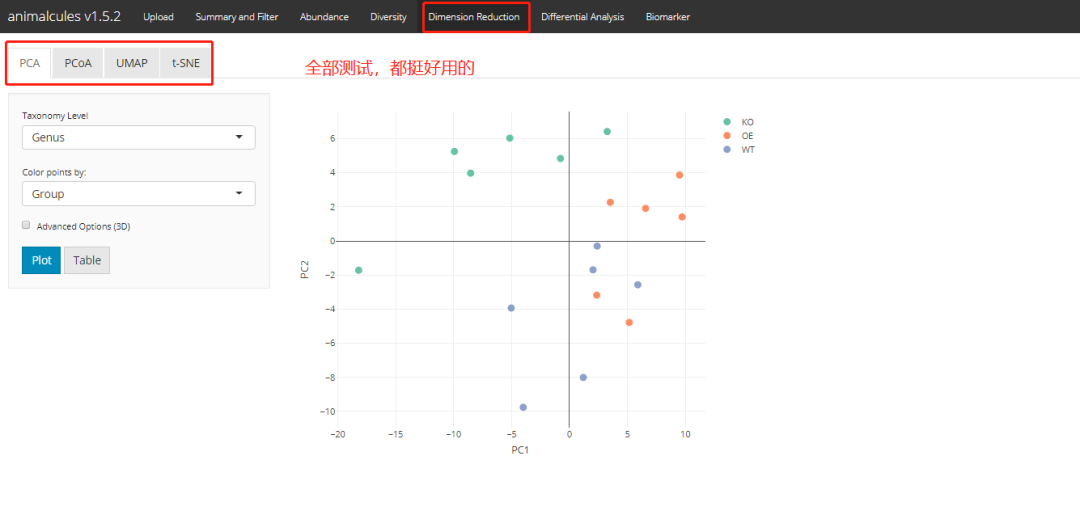
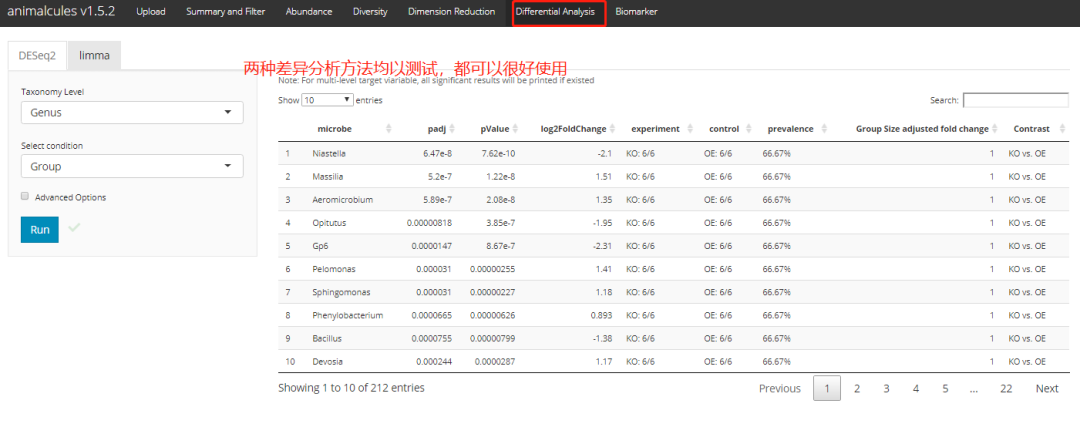
差异分析
p <- differential_abundance(MAE,
tax_level="phylum",
input_da_condition=c("DISEASE"),
min_num_filter = 2,
input_da_padj_cutoff = 0.5)
pshiny版本:
生物标记物分析
这里可选的方法有两个:”logistic regression”, “random forest”。
# ?find_biomarker
p <- find_biomarker(MAE,
tax_level = "genus",
input_select_target_biomarker = c("SEX"),
nfolds = 3,
nrepeats = 3,
seed = 99,
percent_top_biomarker = 0.2,
model_name = "logistic regression")
p$biomarker对重要变量可视化。
# importance plot
p$importance_plot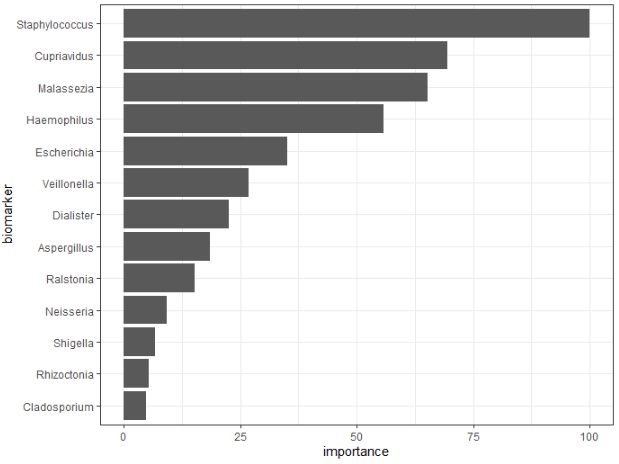
ROC曲线准确度评估。注意ROC曲线只能对二分变量进行操作。
p$roc_plot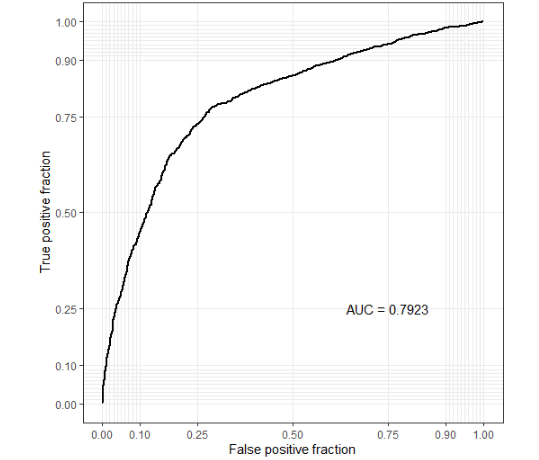
shiny版本(error):
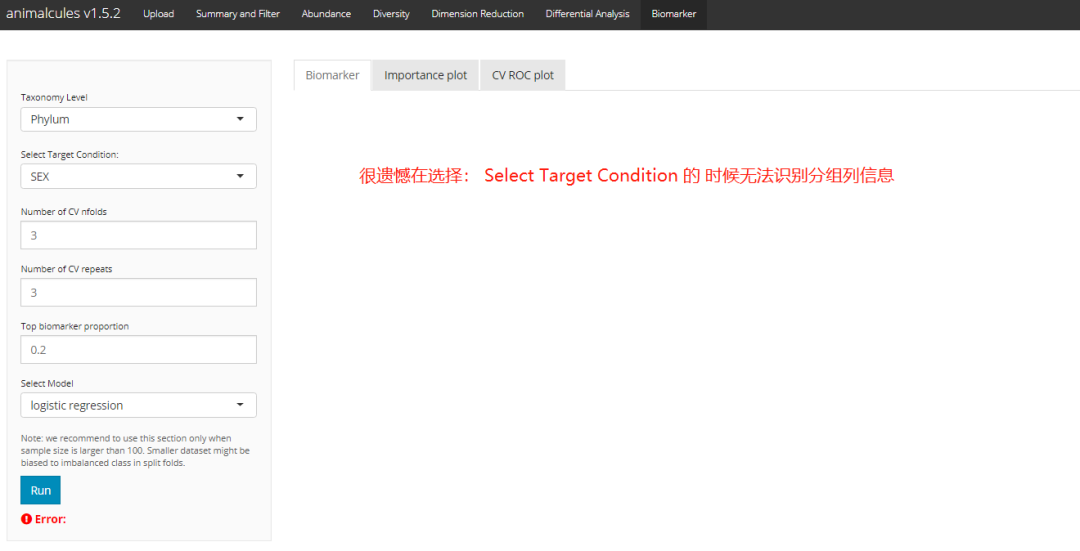
运行环境
sessionInfo()R version 4.1.0 (2021-05-18)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19042)
Matrix products: default
locale:
[1] LC_COLLATE=Chinese (Simplified)_China.936
[2] LC_CTYPE=Chinese (Simplified)_China.936
[3] LC_MONETARY=Chinese (Simplified)_China.936
[4] LC_NUMERIC=C
[5] LC_TIME=Chinese (Simplified)_China.936
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] caret_6.0-88 biomformat_1.20.0
[3] magrittr_2.0.1 dplyr_1.0.6
[5] vegan_2.5-7 lattice_0.20-44
[7] permute_0.9-5 plotly_4.9.3
[9] ggplot2_3.3.3 shinyjs_2.0.0
[11] shiny_1.6.0 phyloseq_1.36.0
[13] ggClusterNet_0.1.0 MultiAssayExperiment_1.18.0
[15] SummarizedExperiment_1.22.0 Biobase_2.52.0
[17] GenomicRanges_1.44.0 GenomeInfoDb_1.28.0
[19] IRanges_2.26.0 S4Vectors_0.30.0
[21] BiocGenerics_0.38.0 MatrixGenerics_1.4.0
[23] matrixStats_0.58.0 animalcules_1.9.1
loaded via a namespace (and not attached):
[1] utf8_1.2.1 reticulate_1.20 tidyselect_1.1.1
[4] RSQLite_2.2.7 AnnotationDbi_1.54.0 htmlwidgets_1.5.3
[7] ranger_0.12.1 grid_4.1.0 BiocParallel_1.26.0
[10] covr_3.5.1 pROC_1.17.0.1 devtools_2.4.2
[13] munsell_0.5.0 codetools_0.2-18 DT_0.18
[16] umap_0.2.7.0 rentrez_1.2.3 withr_2.4.2
[19] colorspace_2.0-1 knitr_1.33 rstudioapi_0.13
[22] labeling_0.4.2 GenomeInfoDbData_1.2.6 plotROC_2.2.1
[25] bit64_4.0.5 farver_2.1.0 rhdf5_2.36.0
[28] rprojroot_2.0.2 vctrs_0.3.8 generics_0.1.0
[31] ipred_0.9-11 xfun_0.23 R6_2.5.0
[34] locfit_1.5-9.4 rex_1.2.0 bitops_1.0-7
[37] rhdf5filters_1.4.0 cachem_1.0.5 DelayedArray_0.18.0
[40] assertthat_0.2.1 promises_1.2.0.1 scales_1.1.1
[43] nnet_7.3-16 gtable_0.3.0 processx_3.5.2
[46] timeDate_3043.102 rlang_0.4.11 genefilter_1.74.0
[49] splines_4.1.0 lazyeval_0.2.2 ModelMetrics_1.2.2.2
[52] GUniFrac_1.2 BiocManager_1.30.15 yaml_2.2.1
[55] reshape2_1.4.4 crosstalk_1.1.1 httpuv_1.6.1
[58] tools_4.1.0 lava_1.6.9 usethis_2.0.1
[61] ellipsis_0.3.2 jquerylib_0.1.4 RColorBrewer_1.1-2
[64] proxy_0.4-26 sessioninfo_1.1.1 Rcpp_1.0.6
[67] plyr_1.8.6 progress_1.2.2 zlibbioc_1.38.0
[70] purrr_0.3.4 RCurl_1.98-1.3 ps_1.6.0
[73] prettyunits_1.1.1 rpart_4.1-15 openssl_1.4.4
[76] cluster_2.1.2 fs_1.5.0 data.table_1.14.0
[79] RSpectra_0.16-0 pkgload_1.2.1 hms_1.1.0
[82] mime_0.10 evaluate_0.14 xtable_1.8-4
[85] XML_3.99-0.6 shape_1.4.6 testthat_3.0.2
[88] compiler_4.1.0 tibble_3.1.2 crayon_1.4.1
[91] htmltools_0.5.1.1 mgcv_1.8-35 later_1.2.0
[94] tidyr_1.1.3 geneplotter_1.70.0 lubridate_1.7.10
[97] DBI_1.1.1 MASS_7.3-54 Matrix_1.3-3
[100] ade4_1.7-16 cli_2.5.0 gower_0.2.2
[103] igraph_1.2.6 forcats_0.5.1 pkgconfig_2.0.3
[106] recipes_0.1.16 foreach_1.5.1 annotate_1.70.0
[109] bslib_0.2.5.1 multtest_2.48.0 XVector_0.32.0
[112] prodlim_2019.11.13 stringr_1.4.0 callr_3.7.0
[115] digest_0.6.27 tsne_0.1-3 Biostrings_2.60.0
[118] rmarkdown_2.8 curl_4.3.1 lifecycle_1.0.0
[121] nlme_3.1-152 jsonlite_1.7.2 Rhdf5lib_1.14.0
[124] desc_1.3.0 viridisLite_0.4.0 askpass_1.1
[127] limma_3.48.0 fansi_0.5.0 pillar_1.6.1
[130] KEGGREST_1.32.0 fastmap_1.1.0 httr_1.4.2
[133] pkgbuild_1.2.0 survival_3.2-11 glue_1.4.2
[136] remotes_2.4.0 png_0.1-7 iterators_1.0.13
[139] glmnet_4.1-1 bit_4.0.4 class_7.3-19
[142] stringi_1.6.2 sass_0.4.0 blob_1.2.1
[145] DESeq2_1.32.0 memoise_2.0.0 e1071_1.7-7
[148] ape_5.5Reference
-
https://bioconductor.org/packages/release/bioc/vignettes/animalcules/inst/doc/animalcules.html
reference:
Interactive microbiome analysis toolkit • animalcules (compbiomed.github.io)
Zhao, Yue, Anthony Federico, Tyler Faits, Solaiappan Manimaran, Daniel Segrè, Stefano Monti, and W. Evan Johnson. "animalcules: interactive microbiome analytics and visualization in R." Microbiome 9, no. 1 (2021): 1-16.