Predicting miRNA-Disease Associations Based On Multi-View Variational Graph Auto-Encoder With Matrix Factorization
![]()
Availability and implementation: The code and datasets of VGAMF are available at: https://github.com/XYDBCS/VGAMF.
Abstract
摘要: 小 RNA (miRNA)已被证明在包括人类疾病发展过程在内的多种生物学过程中发挥着重要作用。探索 miRNA 与疾病之间的潜在联系可以帮助我们更好地理解复杂的疾病机制。考虑到传统的生物实验是昂贵和耗时的,计算模型可以作为发现潜在的 miRNA 疾病关联的有效手段。这项研究提出了一种新的基于带有矩阵分解的变分图自动编码计算模型(VGAMF),用于 miRNA-疾病关联预测。更具体地说,VGAMF 首先将关于 miRNA 的四种不同类型的信息分别集成到 miRNA 综合相似性网络和关于疾病的两种类型的信息集成到疾病综合相似性网络中。然后,VGAMF 从这两个具有变分图自动编码器的综合相似性网络中分别获得 miRNA 和疾病的非线性表征。同时,在 miRNA-疾病关联矩阵上进行一个非负矩阵分解,以获得 miRNA 和疾病的线性表征。最后,一个全连接的神经网络将 miRNA 和疾病的线性和非线性表征结合起来,得到所有 miRNA-疾病对的最终预测关联评分。在10折的交叉验证实验中,VGAMF 在 HMDD v2.0上达到了0.9280的平均 AUC,在 HMDD v3.2上达到了0.9470,这比其他竞争方法都要好。此外,结肠癌和食管癌的案例研究进一步证明了 VGAMF 在预测新的 miRNA-疾病关联方面的有效性。
目录
1.引言
现有的方法虽然取得了很大的进展,但都存在一些局限性:首先,将多视图相似性合理地集成到一个综合的相似性网络中是一个挑战。大多数模型通过使用一个相似度来组合不同层次的相似度,从而填补另一个相似度或不同类型相似度的平均值的缺失部分。然而,不同的规则根据不同的证据得到不同的相似性。当使用不恰当的融合方法时,多重相似性中的尺度差异、收集偏差和噪声(scale, collection bias, and noise)可能导致综合相似性网络的质量较差。 在这种情况下,虽然多视图证据可以为预测模型提供更多的信息,但噪声不可避免地影响预测的准确性。其次,一些基于相似性网络的方法和基于矩阵补全的方法严重依赖于现有的 MDA 信息,如果没有已知的 miRNA,就不能应用于新的疾病。第三,对于一些监督式学习方法,最终模型严重受到训练样本质量的影响,而目前的数据库中没有经过验证的阴性样本。随机选取未知样本作为阴性样本可能会降低模型训练的质量。最后,将相似网络和关联矩阵中的所有信息集成起来,很难获得适合于 miRNA 和疾病的特征表示。一些方法通过提取 miRNA 和疾病的线性特征来预测 MDA,而另一些方法则提取深度非线性关联。线性特征和深度非线性特征各有优势,但很少有方法同时具备这两种特征。
在这项研究中,为了解决上面提到的一些局限性,我们提出了一种新的基于带有矩阵分解的变分图自动编码器(VGAMF)的用于 MDA 预测的计算模型。VGAMF 首先通过整合不同的数据库,分别计算出 miRNA 和疾病的两个综合相似性网络。具体而言,VGAMF 构建了包括 miRNA 序列相似性、功能相似性、语义相似性和高斯互作谱核相似性在内的四个 miRNA 相似性网络,以及包括疾病语义相似性和高斯互作谱核相似性在内的两个疾病相似性网络。然后,通过非线性融合方法将这些不同的相似性网络分别集成到 miRNA 和疾病的综合相似性网络中。然后,在两个综合相似网络上分别训练两个变分图自动编码器,得到 miRNA 和疾病的非线性表征。同时,在 MDA 矩阵上执行非负矩阵分解(NMF)模型以提取线性表征。最后,一个全连接的神经网络将 miRNA 和疾病的线性和非线性表征结合起来,得到每个 miRNA-疾病对的最终关联预测评分。与以往的 MDA 预测方法相比,VGAMF 使用非线性相似性网络融合方法从不同的数据源获取共享和互补的信息[42]。VGAE 模型可以很好地提取 miRNA 和疾病的深度,复杂的特征,因为 GCN 可以自然地结合图结构中的节点特征,而变分自动编码器(VAE)可以从数据分布的角度捕获特征。此外,线性表示和非线性表示的结合为最终的预测提供了更多来自不同层次的信息。
2.材料和方法
在第 II-A 节中,描述了模型中涉及的数据源。在第 II-B 节中,介绍了基于各种数据源分别计算 miRNA 和疾病相似性的不同方法。融合方法是一种将不同的相似性融合到一个综合相似性网络中的方法。在第 II-C 部分,VGAE 被提出用于获得 miRNA 和疾病的非线性表征。此外,本文还总结了利用 NMF 获得 miRNA 和疾病的线性表征的过程。最后,在第二章 E 节中介绍了 VGAMF 的全过程。
2.A.基准数据集
在这项研究中,我们在两个 miRNA 疾病关联数据集上证明了我们的模型的表现,常用版本 HMDD v2.0[16]和最新版本 HMDD v3.2[43]。HMDD v2.0和相关的相似性数据集是从以前的研究[44]中获得的,而 HMDD v3.2和相关的相似性数据集是根据与研究[44]中相同的过程预处理的。具体而言,原始的 HMDD v2.0数据库包括从 PubMed 中所有 miRNA 相关出版物手动收集的577个 miRNA 和366个疾病之间的6441个关联。为了在 VGAMF 中集成更多的信息,使用了一些数据库进行相似度计算。首先,miRNA 序列信息来自 miRBase [45] ,包含4796个人类 miRNA 注释信息。MiRNA 相关的基因信息来自 mirTarBase [46] ,其中包括2599miRNA 和15064个基因之间的380693个相互作用。疾病语义信息来自国家医学图书馆( http://www.nlm.nih.gov/)最新的 MeSH 描述符。它包括11572个来自 C 类疾病的独特条目。为了保持来自不同来源的数据的一致性,在用 miRBase 记录、 mirTatBase 记录和 MeSH 描述符映射 HMDD v2.0中的 miRNA 名称和疾病名称之后,最终保留了550个 miRNA 和328种疾病之间的6088个关联,构建了一个表达所有 MDAs 的关联矩阵 ![]() 。如果 miRNA i 和疾病 j 相关,则
。如果 miRNA i 和疾病 j 相关,则 ![]() = 1,否则
= 1,否则 ![]() = 0。
= 0。
此外,原始的 HMDD v3.2[43]在去除重复关系后,包含1208个 miRNA 和894个疾病之间的18733个关联。我们删除 HMDD v3.2中但不存在于数据库 miRBase 或 mirTarBase 中的 miRNA,并删除 HMDD 3.2中但不存在于 MeSH 中的疾病。然后,将788个 miRNA 与374种疾病之间的8968个关联留下来构建一个关联矩阵 ![]() 。在这两种类型的数据库中,相似网络是按照以下过程计算的。
。在这两种类型的数据库中,相似网络是按照以下过程计算的。
2.B.相似性网络
疾病相似性测量。为了得到一个全面的疾病相似性矩阵,我们采用两种不同的标准来评价疾病-疾病的相似性。
1)疾病语义相似性。MeSH 描述符用于实现疾病的有向无环图(DAGs)。基于这样的假设,即两种疾病的 DAGs 大部分是共同的,两种疾病在语义上更相似,我们使用以前的研究[47]中的方法来计算两种疾病之间的语义相似性。因此,我们得到了一个疾病语义相似度矩阵,表示为 HMDD v2.0的 ![]() ,或者 HMDD v3.2的
,或者 HMDD v3.2的 ![]() 。
。
2)疾病高斯互作谱(GIP)核相似性。根据从 HMDD 获得的 MDA 矩阵 A 计算疾病 GIP 核相似性,假设与相同 miRNA 相关的疾病更可能相似,反之亦然。疾病 GIP 核相似性由以前的方法[48]计算,疾病 GIP 核相似性矩阵由 HMDD v2.0的 ![]() 或 HMDD v3.2的
或 HMDD v3.2的 ![]() 表示。.
表示。.
MiRNA 相似性测量。为了全面描述 miRNA 之间的相似性,我们采用四种不同的标准来评估 miRNA-miRNA 的相似性。
1) miRNA 序列相似性。MiRNA 的序列来自数据库 miRBase,对于每个 miRNA,整个成熟序列约为22个核苷酸“ AUCG”。基于序列信息,我们使用 R 软件包 Biostring 中的成对序列比对函数“ pairwiseAlignment”来计算相似度得分。在该函数中,间隙开放惩罚设置为5,间隙扩展惩罚设置为2,匹配得分设置为1,错配得分设置为 -1。在得到序列相似性评分后,我们使用最大-最小归一化方法将其归一化到范围[0,1]。最后,我们得到了 HMDD v2.0的 miRNA 序列相似矩阵 ![]()
![]() ,HMDD v3.2的 miRNA 序列相似矩阵
,HMDD v3.2的 miRNA 序列相似矩阵 ![]() 。
。
2) miRNA 功能相似性。基于功能相似的 miRNA 更可能与相同疾病相关的假设,根据以前的方法,通过其相关疾病 DAGs 的相似性来测量 miRNA 的功能相似性[47]。MDA 来自 HMDD,疾病 DAGs 根据 MeSH 描述符构建。最后,我们得到了表示为 HMDD v2.0的 ![]() 或 HMDD v3.2的
或 HMDD v3.2的![]() 的 miRNA 功能相似性矩阵。
的 miRNA 功能相似性矩阵。
3) miRNA 语义相似性。通过 miRNA 靶基因和基因相关的本体(GO)注释描述 miRNA 的语义相似性。MiRNA 靶基因信息来自 mirTarBase。对于每对 miRNA,我们获取它们的目标基因列表,然后通过以前研究中的方法计算两个相应基因组之间的语义相似性[49]。类似地,我们得到了一个 miRNA 语义相似矩阵,表示为 ![]() (HMDD v2.0)或
(HMDD v2.0)或 ![]() (HMDD v3.2)。
(HMDD v3.2)。
4) miRNA GIP 核相似性。基于与疾病 GIP 核相似性计算相同的方法,如前面的方法[48]计算 miRNA GIP 核相似性矩阵,HMDD v2.0的 miRNA GIP 核相似性矩阵为 SM4 something R550 * 550,HMDD v3.2的 miRNA GIP 核相似性矩阵为 R788 * 788。注意,在交叉验证中,在计算每折的 GIP 核相似度之前,测试样本中的正相关应该在 miRNA-disease 关联矩阵中设置为未知。
将不同的相似性融合到一个综合的相似性网络中。 受以前研究[42]的启发,我们使用非线性融合方法将各种相似性测量分别整合到 miRNA 和疾病的单一相似性网络中。与大多数简单的线性相似性组合方法相比,该方法能够很好地捕获来自不同数据源的共享信息和互补信息,具有抗噪性和数据异构性。本文以全面的 miRNA 相似性为例进行了构建。
首先,我们对每种类型的相似网络进行了更好的归一化。以 miRNA 序列相似性矩阵 SM1为例,计算重归一化过程如下:

这种规范化不受对角线条目自相似性的影响,每一行的总和仍然是1。
然后,对于一定的相似网络 GM,如序列相似网络 SM1,我们使用 K 最近邻(KNN)来度量其局部亲和度 S _ kn 如下:

其中 Ni 是一组最接近 xi 的 K,包括 G 中的 xi。该算法基于局部相似度(高相似度值)比远程相似度更可靠的假设,并将与远程相似度设置为0。
在得到与各类数据相对应的局部亲和核之后,我们对每类数据的相似矩阵进行迭代更新如下:

其中 m 是数据类型的总数,v 是当前数据类型的数量,范围从1到 m,![]() 表示经过 t 次迭代后第 v 数据类型的状态矩阵,
表示经过 t 次迭代后第 v 数据类型的状态矩阵,![]() 表示 vth 数据类型的局部亲和核。
表示 vth 数据类型的局部亲和核。
每次迭代后,对状态矩阵 ![]() 进行归一化处理如方程(1)。当迭代达到收敛准则时,即当相对变化量
进行归一化处理如方程(1)。当迭代达到收敛准则时,即当相对变化量  小于10-6时,迭代停止。当每种类型的数据的迭代停止时(我们假设它包括 t 次迭代) ,总体综合相似性矩阵计算如下:
小于10-6时,迭代停止。当每种类型的数据的迭代停止时(我们假设它包括 t 次迭代) ,总体综合相似性矩阵计算如下:

根据上述规则,在方程中的相似矩阵。(4)不是一个对称矩阵,所以我们计算 ![]() 作为 miRNA 的综合相似度矩阵。对于疾病,我们遵循与 miRNA 相同的规则,得到疾病综合相似度矩阵 SD。
作为 miRNA 的综合相似度矩阵。对于疾病,我们遵循与 miRNA 相同的规则,得到疾病综合相似度矩阵 SD。
2.C.VAGE获取非线性表示
VGAE 是一个无监督式学习模型,它结合了 GCN 和变分自动编码器(VAE)。该模型通常应用于图结构数据。利用 VAE 的潜在变量和 GCN 的邻域信息融合能力,学习无向图的可解释潜表征。VGAE 获得的非线性表示可以集成图结构和数据分布。接下来,我们将详细介绍 GCN 和 VGAE。
GCN [50]是针对具有非欧几里德数据的底层图结构的卷积运算而提出的。近年来,GCN 为许多基于网络的预测任务带来了显著的性能改进,例如 lncRNA 疾病关联预测[51]和 miRNA 耐药性预测[52]。GCN 通过集成相邻节点信息和图结构信息,可以有效地学习图中每个节点的特征向量。目前,基于对局部卷积滤波器不同定义的 GCN 方法分为两类: 一类是基于空间的方法,另一类是基于谱的方法。正如 Bruna 的研究[53]中提到的,基于谱的方法是基于图 Laplacian [50]的谱设计的,它通常比基于空间的方法具有更好的性能,这些方法具有许多局限性。因此,本研究采用基于谱的方法,分别从 miRNA 相似性网络和疾病相似性网络中提取 miRNA 和疾病特征向量。
让相似矩阵 SM 成为 ![]() miRNAs 的邻接矩阵。我们将 miRNA X 的初始标量特征设置为 miRNA 疾病邻接矩阵的一行。在获得输入数据 SM 和 X 之后,GCN 通过以下公式将步骤 t-1处的图信号 X (t-1)转换成新的信号 X (t):
miRNAs 的邻接矩阵。我们将 miRNA X 的初始标量特征设置为 miRNA 疾病邻接矩阵的一行。在获得输入数据 SM 和 X 之后,GCN 通过以下公式将步骤 t-1处的图信号 X (t-1)转换成新的信号 X (t):

其中 ![]() 是一个矩阵,它将 SM 的所有对角线元素设置为1,表示 GM 的自循环邻接矩阵,
是一个矩阵,它将 SM 的所有对角线元素设置为1,表示 GM 的自循环邻接矩阵,![]() 是一个度对角线矩阵,其中
是一个度对角线矩阵,其中 ,
,![]() 是 GCN 模型(t-1)层中的参数,而 Relu ()是一个非线性激活函数,它也可以被 sigmoid ()或其他一些激活函数替换。
是 GCN 模型(t-1)层中的参数,而 Relu ()是一个非线性激活函数,它也可以被 sigmoid ()或其他一些激活函数替换。
我们的模型结合了 GCN 和变分自动编码器(VAE)作为 VGAE 来提取 miRNA 和疾病的非线性表示。在 VGAE,GCN 可以在网络中加入节点特征,而 VAE 使用潜在变量从数据分布的角度学习可解释的潜在表征。
VGAE 包括编码器和解码器。在编码部分,它以一个邻接矩阵的 SM 和一个特征矩阵 X 作为输入,通过GCN得到一个潜变量 z 作为输出,而在解码部分,VGCN 基于潜变量 z 重建邻接矩阵 SM。它还包括一个损失函数,用于获得最优参数。
编码器:编码器包括两层 GCN。第一 GCN 层生成低维特征矩阵。其定义如下:

第二个 GCN 网络层产生如下数据分布:

然后,潜变量 z 计算如下:
![]()
其中 ε 遵循标准正态分布 N (0,1)。编码器也可以表示如下:
![]()
译码器:译码器由潜变量 z 之间的内积定义,输出是一个重构的邻接矩阵,如下所示:
![]()
其中 S 是 sigmoid 函数。
译码器也可以表示如下:
![]()
损失函数:损失函数包括两部分。第一部分是目标 SM 与输出 SM 之间的二元交叉熵,第二部分是 ![]() 与 p (Z)之间的 KL- 散度。损失函数的定义如下:
与 p (Z)之间的 KL- 散度。损失函数的定义如下:

VGAE 在 miRNA-miRNA 相似性网络上提取 miRNA 的非线性表示的整个过程如图1所示。潜变量矩阵 Z 被认为是 miRNA 的非线性表示![]() 。相似的,我们输入疾病-疾病相似性矩阵 SD 作为邻接矩阵,MDA 矩阵 A 的每一列作为初始疾病特征,并得到非线性疾病表示为
。相似的,我们输入疾病-疾病相似性矩阵 SD 作为邻接矩阵,MDA 矩阵 A 的每一列作为初始疾病特征,并得到非线性疾病表示为 ![]() 。
。

2.D.NMF获取线性表示
非线性表示主要来自综合相似网络。虽然它们整合了多视图数据集来接收更多信息,但是它们也包含一些噪声,因为所有这些相似性都是基于不同的度量计算的。在这一部分中,miRNA 和疾病的线性表示由基于 MDA 矩阵的 NMF 计算。
NMF 将 miRNA-疾病关系投射到 miRNA 子空间和疾病子空间中,通过将原始 MDA 矩阵分解成两个低秩矩阵来帮助揭示潜在特征,并使其相乘尽可能近似原始矩阵[54]。假设 MDA 矩阵 ![]() 与低秩 miRNA 特征矩阵
与低秩 miRNA 特征矩阵 ![]() 和疾病特征矩阵
和疾病特征矩阵 ![]()
![]() 的内积非常接近,[54] m 是 miRNA 的个数,n 是疾病的个数,k 是特征空间维数。为了充分利用已验证的关联,减少未知关联的不利影响,提出了一个指示器加权矩阵
的内积非常接近,[54] m 是 miRNA 的个数,n 是疾病的个数,k 是特征空间维数。为了充分利用已验证的关联,减少未知关联的不利影响,提出了一个指示器加权矩阵 ![]() 。W的值和A一样。此外,我们使用
。W的值和A一样。此外,我们使用 ![]() 正则化[55]来保证 U 和 V 的光滑性,然后定义目标函数如下:
正则化[55]来保证 U 和 V 的光滑性,然后定义目标函数如下:

其中 λ1和 λ2是正则化系数,![]() 是 Hadamard 乘积。U ≥0(V ≥0)意味着 U (V)的所有条目都是非负的。
是 Hadamard 乘积。U ≥0(V ≥0)意味着 U (V)的所有条目都是非负的。
设 ![]() 是拉格朗日乘子,即优化问题方程的拉格朗日函数。(15)可按以下方式构造:
是拉格朗日乘子,即优化问题方程的拉格朗日函数。(15)可按以下方式构造:
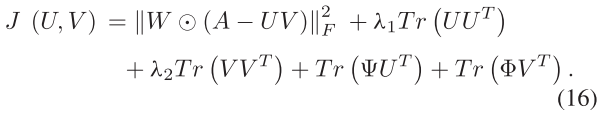
J 的偏导数计算如下:

根据 Karush-Kuhn-Tucker (KKT)条件[56] ,![]() ,我们可以得到
,我们可以得到 ![]() 的乘法更新规则如下:
的乘法更新规则如下:

根据方程(19)及方程(20) 中的更新规则,当相对差收敛准则达到10-4时,我们可以得到 U 和 V。
2.E.VGAMF预测 miRNA - 疾病关联
在这一部分,我们介绍了 VGAMF 的整个过程,其中包括五个步骤,如图2所示。
步骤1: 从多视图数据库中,VGAMF 计算 miRNA 的四种不同类型的相似性网络(包括 miRNA 序列相似性,miRNA 功能相似性,miRNA 语义相似性和 miRNA GIP 核相似性)和疾病的两种不同类型的相似性网络(包括疾病语义相似性和疾病 GIP 核相似性)。
步骤2: VGAMF 将这些不同的相似性融合成一个全面的 miRNA 相似性网络 SM 和一个全面的疾病相似性网络 SD。
步骤3: VGAMF 以相似性网络和 miRNA 疾病邻接矩阵的节点特征矩阵作为输入,分别从综合相似性网络 SM 和 SD 中提取 miRNA 和疾病的非线性表示。
步骤4: 基于 miRNA-disease 邻接矩阵,NMF 提取 miRNA 和疾病的线性表示。线性表示只基于 MDA 邻接矩阵。
步骤5: VGAMF 将非线性表示和线性表示相结合,利用完全连通的神经网络进行 MDA 预测。

3.结果与讨论
4.结论
发现潜在的 MDA 可以帮助我们在分子水平上更好地了解疾病的发病机制,提高疾病的诊断、预后和治疗。然而,通过生物学实验揭示 miRNA 与疾病之间的关联是低效的。近年来,随着许多与 miRNA 和疾病相关的数据库的建立,人们提出了各种计算 MDA 的预测方法。本研究整合多视图资料库,提出基于变分图自动编码器及矩阵分解的 MDA 预测方法 VGAMF。采用非线性相似性融合方法,将不同类型的 miRNA 和疾病相关信息分别融合到综合 miRNA 相似性网络和综合疾病相似性网络中。然后使用 VGAE 从 miRNA 和疾病的综合相似性网络中提取深度非线性表示,而使用 NMF 从 miRNA 和疾病的邻接矩阵中提取 miRNA 和疾病的线性表示。结合线性和非线性表示,得到最终的预测关联分数。5折 CV 和10折 CV 的实验结果表明,VGAMF 方法比竞争方法具有更好的预测性能。此外,案例研究也显示了 VGAMF 对预测潜在 MDA 的有效性。
VGAMF 的可靠预测性能主要取决于以下因素。首先,利用非线性融合方法将不同类型的数据库有效地整合到相似性网络中。该融合方法能够同时获取各种数据源的共享信息和互补信息,比线性相似度整合方法有更好的融合效果。其次,VGAMF 通过自然地结合图结构中的节点特征,利用潜在变量从数据分布的角度对数据进行分析,可以有效地从网络中提取信息。第三,线性表征与非线性表征相辅相成。非线性表示主要是基于相似网络提取的,其中包含有噪声的多视点信息,而线性表示只是基于可靠验证的 MDA 矩阵。此外,一个表示是通过深入的非线性过程提取的,而另一个表示浅层线性关系。最后,VGAMF 可以减轻随机选择的阴性样本噪声的影响。
然而,VGAMF 也有一些局限性,需要进一步的研究。例如,疾病相似性仅包括两种信息。今后,我们将在疾病相似性网络中整合更多与疾病相关的证据。此外,VGAMF 中的非线性表示严重依赖于含有噪声的相似网络的质量。在未来,我们将进一步研究一种能够有效利用 miRNA 和疾病的多视图信息的方法,同时尽可能减少噪声信息。此外,我们会考虑更多的生物相关分析,以说明我们的预测模型的有效性,如生存分析或药物敏感度分析。